

Prof Thomas Milne, Dr Nicholas Crump, Dr Anindita Roy, Dr Laura Godfrey
Image: Abstract spray painting to represent identification of the right molecular target in a complex leukemia (credit: Adi Sukumaran)
One of the things that we get used to saying when writing about the Mixed Lineage Leukemia (MLL) gene is that mutations in this gene cause very poor prognosis leukemias in children and adults and thus remain an unmet need. It is easy to forget that behind this blanket scientific statement are real individuals, and their families, who face this disease with very few curative treatment options. Unfortunately, the current excitement over immunotherapies is not applicable to MLL leukemias, as they often relapse after treatment with Chimeric Antigen Receptor (CAR) T cell therapies. It has become a cliché to say that better therapies are needed. But how can we understand the leukemia better in order to deliver them?
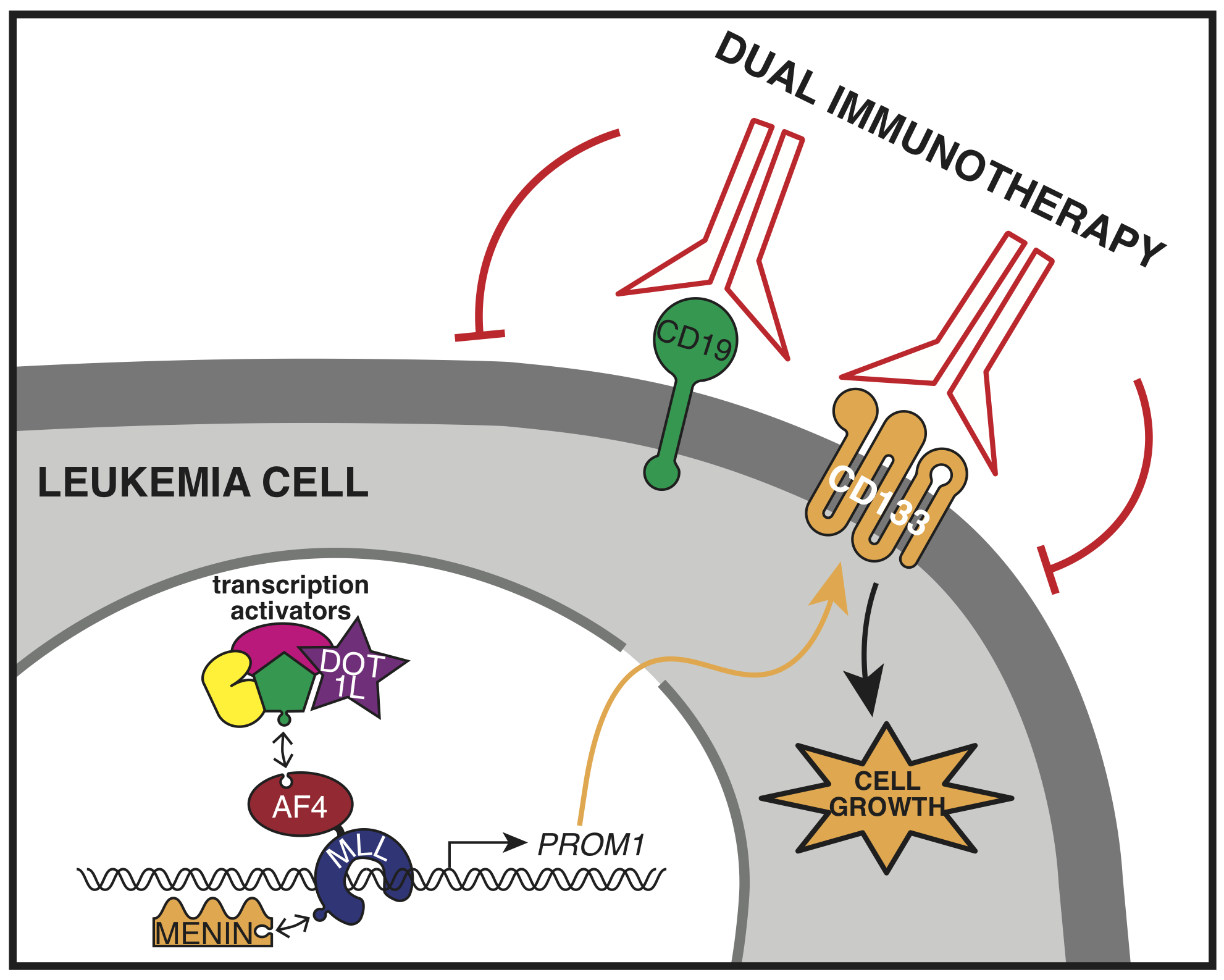
The most common MLL gene mutation is a chromosome rearrangement that fuses it with the AF4 gene creating MLL-AF4, a new protein that shouldn’t normally exist, and is capable of completely reprogramming gene expression in the cell. The MLL-AF4 protein drives leukemia by binding to and activating genes that do things such as promote growth and block cell death. So far, it has been very difficult to develop drugs targeting the MLL-AF4 protein complex itself to block its activity, although some progress in this area has been made with inhibitors to the H3K79 methyltransferase DOT1L and more recently the protein Menin. However, an alternative strategy is to target the protein products of the activated genes. One of these is PROM1.
Interestingly, like many scientific discoveries, we stumbled across the PROM1 gene almost by accident. While trying to better understand the activity of the MLL-AF4 protein we treated leukemia cells with a DOT1L inhibitor and found that the most highly disrupted gene was PROM1. We hadn’t heard of PROM1 before, but soon found out that it encoded the cell surface receptor CD133, which has long been associated with MLL leukemias. It’s widely expressed on many cancer stem cells as well as normal stem cells, but little work had been done on the regulation of the gene itself.
At the time, we were exploring the role of the histone modification H3K79me2/3 in enhancer activity, and it seemed natural to include PROM1/CD133 in our analysis in MLL-AF4 leukemia cells. As we began to study its regulation in more detail, we found that PROM1/CD133 gene expression is controlled by a balance between the Polycomb Repressive Complex 2 (PRC2) and the MLL fusion protein, with MLL-AF4 activating two enhancers that drive expression. In MLL leukemias which don’t express PROM1, PRC2 is bound at the promoter instead of the MLL fusion protein. We’re not sure why PROM1 is active in some MLL leukemias but repressed in others, but we think it’s probably a consequence of the specific MLL fusion partner, or the cell that the mutation originally arose in. What we do know is that MLL-AF4 leukemia cells that express PROM1 are absolutely dependent on it, and stop growing if it’s taken away.
This is an exciting finding and provides a potential Achilles’ heel for curing this dreadful leukemia using targeted treatments, including immunotherapy. One of the big problems with treating MLL leukemia with anti-CD19 directed CAR-T cell therapy is that the leukemia cells are able to shut down expression of CD19 and relapse as a resistant cancer. However, our work shows that leukemia cells expressing PROM1/CD133 are addicted to it, and stop growing if it’s inactivated. Therefore, it makes an attractive potential target for dual immunotherapy in combination with CD19 targeting, and that’s what we and others are planning to test next!
Follow the Topic
-
Leukemia
This journal publishes high quality, peer reviewed research that covers all aspects of the research and treatment of leukemia and allied diseases. Topics of interest include oncogenes, growth factors, stem cells, leukemia genomics, cell cycle, signal transduction and molecular targets for therapy.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in