Discovery of rare bacteria by turning read mapping positions into frequency signals
Published in Microbiology

Fourier transforms are powerful mathematical operations that we all use whenever efficient signal processing is required – for example, when we use our mobile phones, laptops, or notebooks or listen to digital music. As scientists, we like to avoid reinventing the wheel, and so we applied these powerful transformations to taxonomically differentiate between closely related microbes and identify rare species in shotgun metagenomic sequencing.
How does it work? Imagine there are a few reads aligning to the reference genome of species A (Figure 1). This could indicate that species A was indeed present in the environment of interest. Since the DNA is amplified on a random basis, the reads would be expected to spread across the reference genome (Figure 1, 2). However, species B, a microbe that acquired genes of species A in the past or shares regions of high sequence similarity, may have been present instead. Then, the reads would cluster at the matching sides (Figure 1, 3). By measuring the distance between all possible position combinations of the mapped reads, transforming the information into frequency information, and comparing it with a reference signal of a perfect uniform read distribution, we can identify core and rare species with reduced false discovery and omission rates.
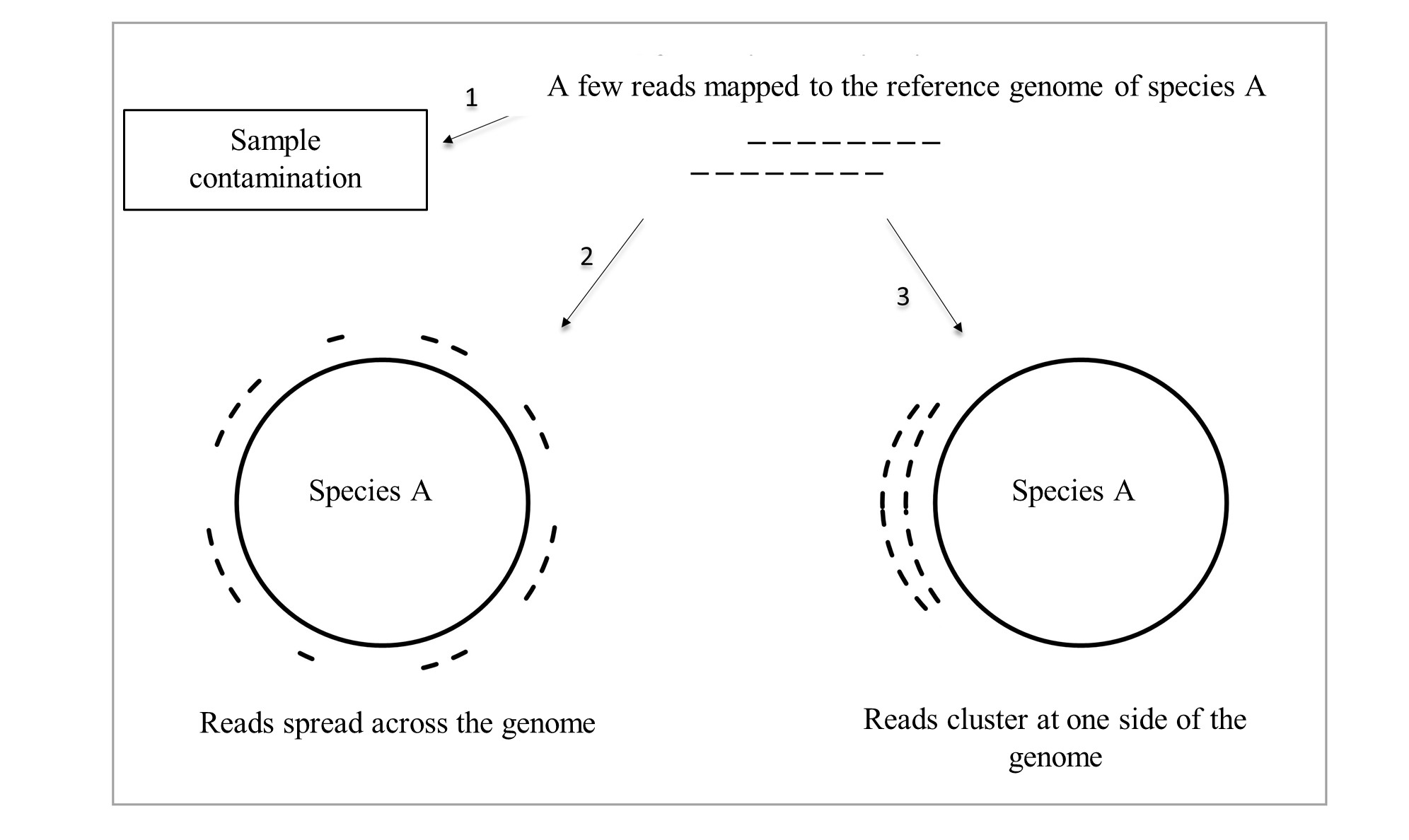
Figure 1. Simplified illustration of read position patterns.
An example may further illustrate the potential use of Fourier transforms. Pseudomonas aeruginosa was cultured in the laboratory, DNA was extracted and sequenced, and the reads were mapped to a reference database. Without Fourier transforms, the true-positive P. aeruginosa, a few false-positive Pseudomonas spp., and Azotobacter vinelandii were detected. A. vinelandii and the Pseudomonas spp. are closely related, and it has been suggested that Azotobacter might actually be a Pseudomonas [1]. After using the information on the distances between read mapping positions to generate frequency signals, we observed a uniform spread of reads along the P. aeruginosa genome (Figure 2). In all other cases, the reference and sample signals were significantly different, revealing the read cluster behaviour and the false-positive nature of these microbes.
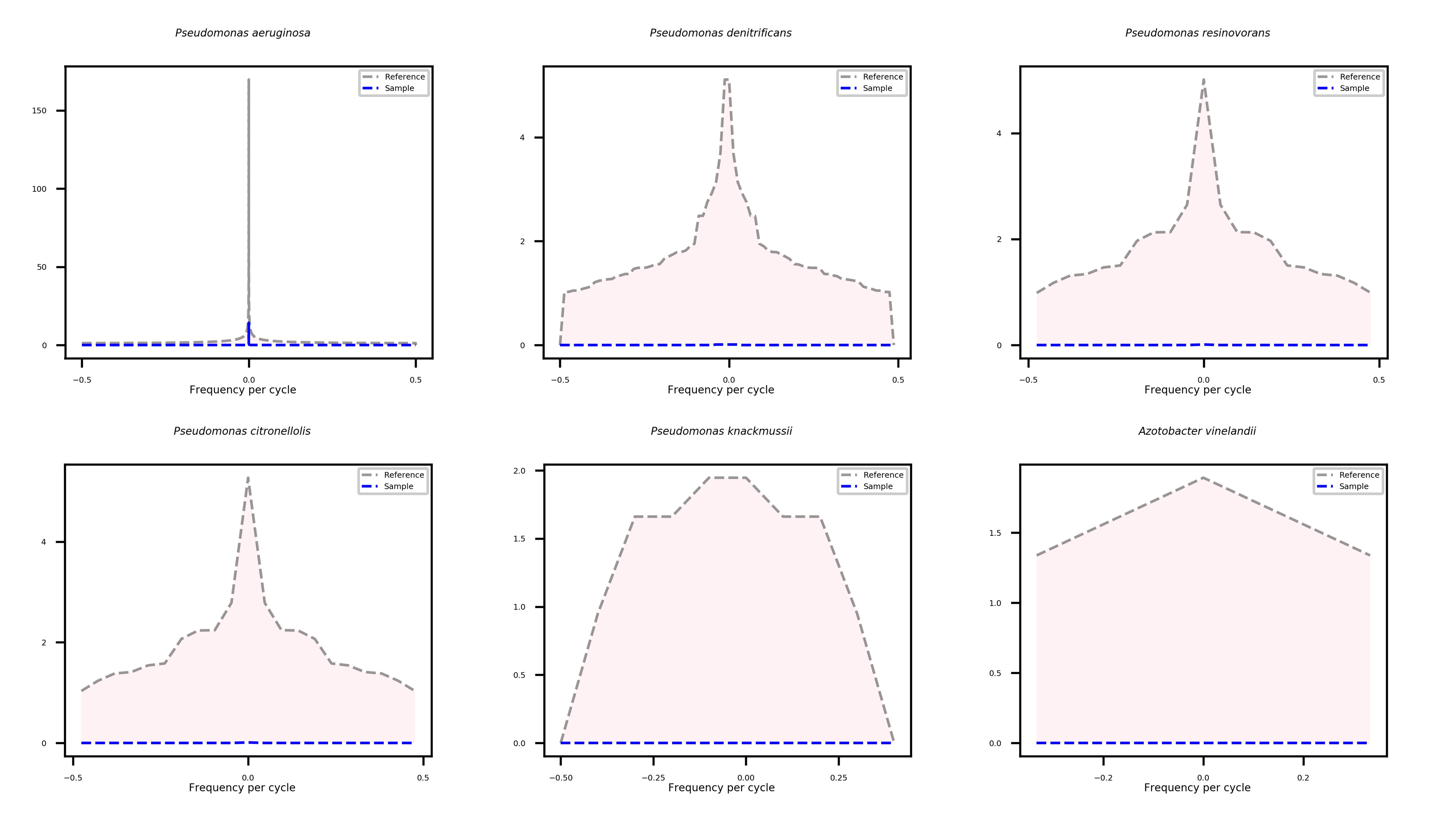
Figure 2. Reference frequency-domain signals (black) and sample signals (blue) obtained for the true-positive P. aeruginosa, four false-positive Pseudomonas spp. (P. resinovorans, P. citronellolis, P. knackmussii), and Azotobacter vinelandii. The red colour depicts the difference between reference and sample signal.
Overall, the number of sequenced reference genomes is on the rise, making it increasingly difficult to differentiate between hundreds and in due course thousands of closely related reference genomes. Our new approach has been tested only on bacteria with circular reference genomes and may not yet be able to solve all the challenging taxonomic classification problems. However, we hope that it will provide a stepping stone for further tools that may rely on Fourier transforms to validate the detection of core and rare fungi, viruses, and circular or non-circular bacteria from various environments.
The article can be accessed via the following link: https://www.nature.com/articles/s43705-021-00010-6
References
[1] Özen AI, Ussery DW. Defining the Pseudomonas genus: where do we draw the line with Azotobacter? Microb Ecol. 2012;63(2):239-248. doi:10.1007/s00248-011-9914-8


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in