Discrete latent embedding of single-cell chromatin accessibility sequencing data for uncovering cell heterogeneity
Published in Genetics & Genomics and Mathematics
Single-cell chromatin accessibility sequencing (scCAS) technologies are flourishing at an unprecedented pace, enabling the interrogation of gene regulation, cell heterogeneity and disease mechanisms. Nonetheless, the intrinsic challenges posed by the high dimensionality, intense sparsity, and nearly binary nature of scCAS data, present formidable obstacles in the realm of data analysis.
What motivated us to develop CASTLE
Recent innovations in computational methods have provided valuable opportunities for capturing low-dimensional embeddings of scCAS data. However, currently employed methods generally bear many shortcomings when modeling and characterizing scCAS data. First, in the variational autoencoder (VAE) models, the Gaussian assumption somewhat disagrees with the discrete nature and the inherent sparsity of scCAS data. On the one hand, the latent space of VAEs gradually approximates the prior distribution, encouraging the cell embeddings gathering near the mean of the latent space and potentially leading to cell crowding. On the other hand, there are many non-Gaussian characteristics in the latent space of a standard VAE model trained on a scCAS dataset, such as multimodality, unexpected skewness and kurtosis. Second, the latent embeddings of existing methods are typically continuous and may not have direct biological implications, making it challenging to quantitatively gain insights into the cell heterogeneity, cell-state transitions, and regulatory mechanisms. Third, recent development of various cell atlases has been making more and more scCAS data available. The valuable information in large-scale existing data is helpful to tackle the high level of noise and technical variations of scCAS data, which suggests the demand for new methods of effectively incorporating massive public scCAS data as reference.
To address these challenges, we develop CASTLE (single-cell Chromatin Accessibility Sequencing data analysis via discreTe Latent Embedding), based on the framework of Vector Quantized Variational AutoEncoder (VQ-VAE)1,2, to interpretably extract discrete latent embeddings and quantitatively generate the cell-type-specific feature spectrum for scCAS data. Moreover, the performance of CASTLE can be further improved by leveraging public unlabeled or labeled reference data via weakly-supervised or supervised manner, respectively.
The framework of CASTLE
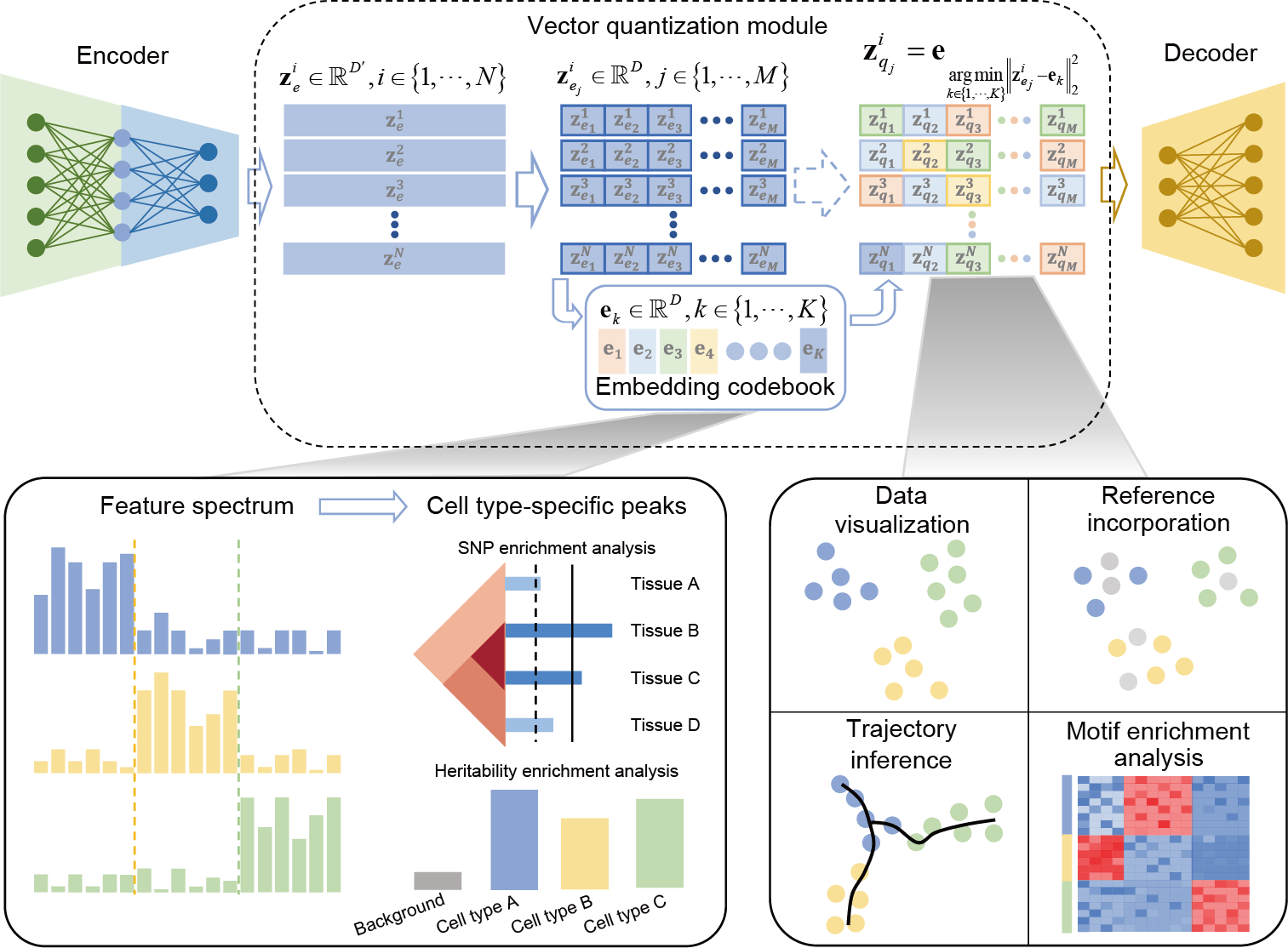
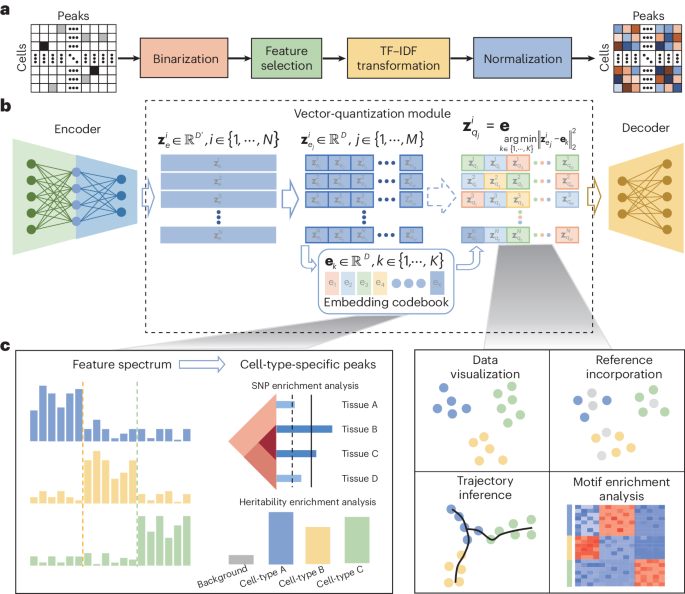
CASTLE takes the processed cell-by-region matrix as input (Fig. 1a). As illustrated in Fig. 1b, CASTLE is composed of three key modules: an encoder, a decoder and a vector quantization module including a discrete embedding space called “codebook”, which can be also learned during the training process. Unlike conventional autoencoder and VAE networks, CASTLE generates the latent quantized feature of a cell by replacing its candidate embeddings from the encoder with the nearest element in the codebook. In other words, the final latent embedding of a cell is composed of vectors in the codebook, which can be considered as a discrete prior distribution that partitions the latent space into independent subspaces. We expected that cells of the same type are mapped into the same code in the codebook, and cells of different types are dispersed into different codes. We further applied split quantization3 to the part of vector quantization to improve the utilization of elements in the codebook. The encoder module is responsible for transforming the input data into the candidate embeddings, whereas the decoder module reconstructs the input data from the quantized features.

Cell type identification with CASTLE
We assessed the capability of CASTLE to identify cell types and uncover cell heterogeneity through clustering. CASTLE outperforms the baseline methods, including cisTopic4, SCALE5, SCALEX6 and scBasset7, by achieving the overall best clustering performance across 16 benchmark datasets (one-sided paired Wilcoxon signed-rank tests P-values < 7e-5) (Fig. 2a). CASTLE achieves a relatively clear separation between HSPC and pDC (highlight with red circle), and distinguishes the rare cell type, CLP (highlight with blue circle) in the UMAP plot (Fig. 2c).
Effective and flexible incorporation of reference data
We extended CASTLE to introduce unlabeled or labeled reference datasets for guiding the cell representation learning process of target datasets. The performance of CASTLE-ref-unlabeled is better than CASTLE without reference datasets, but is worse than CASTLE-ref-labeled (Fig. 2b). Intuitively, when incorporating reference dataset with more information, CASTLE will capture better and more informative cell embeddings for the analysis of scCAS data. Moreover, CASTLE has substantial adaptability to the condition and form of reference datasets.
Feature spectrum for cell heterogeneity uncovering
CASTLE can generate specific feature spectrum for each cell type to reveal biological implications intuitively and quantitatively. Specifically, in the feature spectrum of the Stimulated Droplet dataset, we consistently observe a distinct set of specific features that are enriched or depleted in the feature spectrum of each cell type (Fig. 2d). For instance, in the feature spectra of the Stimulated Droplet dataset, there is a relatively clear difference between the feature spectra of Ery-late and Ery-early while they are connected together in the UMAP visualization (Fig. 2c), suggesting that feature spectrum can unveil cell heterogeneity interpretably. Even the rare cell type, CLP, obtains a set of cell-type-specific features, demonstrating that feature spectrum is robust in revealing cell heterogeneity with different numbers of cells. The cell-type-specific feature spectrum, introduced based on discrete cell embeddings, provides an interpretable approach for illustrating the general and comprehensive patterns of cells belonging to a cell type, further elucidating the cell heterogeneity quantitatively. Furthermore, based on the cell-type-specific peaks identified from cell-type-specific feature spectrum, CASTLE has the great potential of deciphering tissue specificity and gene regulatory mechanisms by SNP enrichment analysis and heritability enrichment analysis.

References:
- van den Oord, A., Vinyals, O. & Kavukcuoglu, K. Neural discrete representation learning. In Proc. 31st Conference on Neural Information Processing Systems 6309–6318 (Curran Associates Inc., 2017).
- Razavi, A., van den Oord, A. & Vinyals, O. Generating diverse high-fidelity images with VQ-VAE-2. In Proc. 33rd Conference on Neural Information Processing Systems 1331 (Curran Associates Inc., 2019).
- Kobayashi, H., Cheveralls, K. C., Leonetti, M. D. & Royer, L. A. Self-supervised deep learning encodes high-resolution features of protein subcellular localization. Nat. Methods 19, 995–1003 (2022).
- Bravo González-Blas, C. et al. cisTopic: cis-regulatory topic modeling on single-cell ATAC-seq data. Nat. Methods 16, 397–400 (2019).
- Xiong, L. et al. SCALE method for single-cell ATAC-seq analysis via latent feature extraction. Nat. Commun. 10, 4576 (2019).
- Xiong, L. et al. Online single-cell data integration through projecting heterogeneous datasets into a common cell-embedding space. Nat. Commun. 13, 6118 (2022).
- Yuan, H. & Kelley, D. R. scBasset: sequence-based modeling of single-cell ATAC-seq using convolutional neural networks. Nat. Methods 19, 1088–1096 (2022).
Follow the Topic
-
Nature Computational Science
A multidisciplinary journal that focuses on the development and use of computational techniques and mathematical models, as well as their application to address complex problems across a range of scientific disciplines.





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in