Dissecting microbiome metabolism based on DNA sequence data
Published in Bioengineering & Biotechnology

Bacteria are indispensable to life on Earth, with a huge impact in humans. As a microbiologist, I have always been fascinated by the different roles microbes play especially in human health and disease and I firmly believe that understanding the way bacteria interact with each other and their environment is fundamental. At early stages, most efforts in the field were made towards assessing the human microbiome composition. These studies showed how patients suffering from several metabolic and immune diseases harbored dysbiotic microbial communities. Bacteria can directly or indirectly promote health or disease by producing hundreds of different metabolites that either interact with host cells or with other microbes. Although cataloging the richness of species of these microbial communities is of relevance, it is important to uncover the molecular mechanisms behind these phenotypes in order to help infer causation. With our new study, we now provide means to analyze the human (or any other animal) microbiome from a different perspective and also describe differences at the mechanistic level that may have an impact on the composition and functioning of this complex ecosystem.
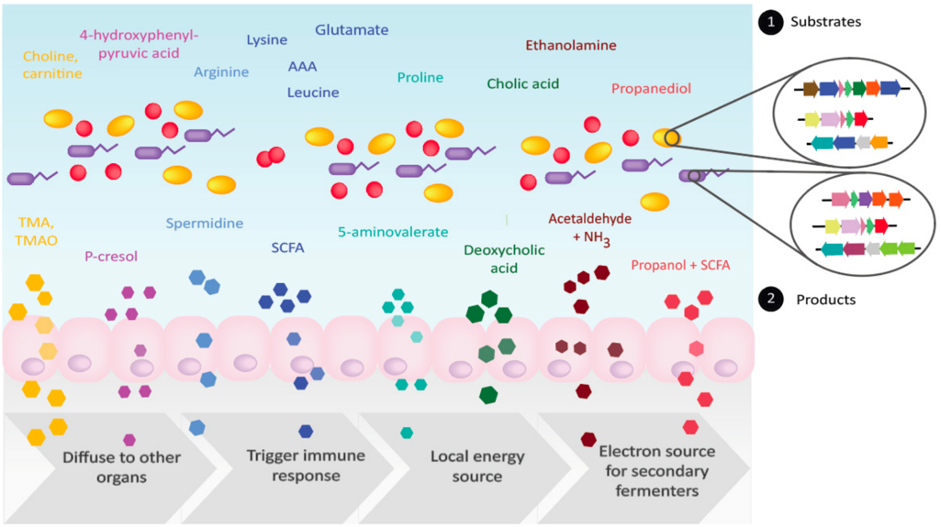
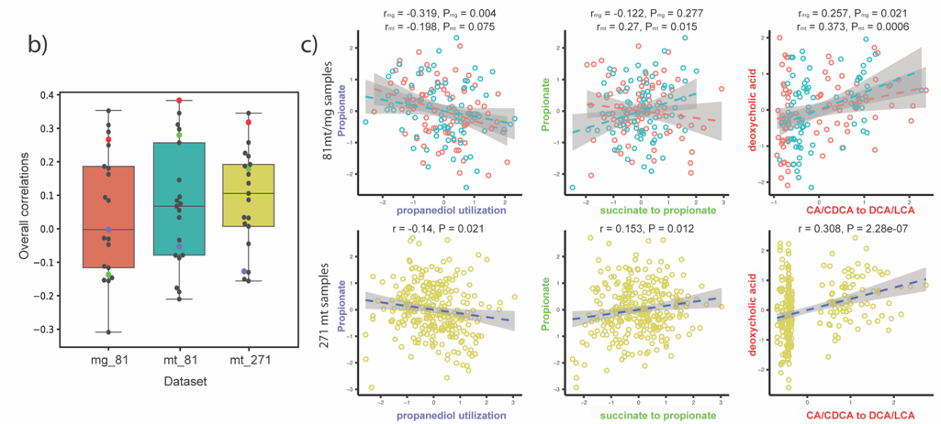
Bacteria in the gut carry out diverse metabolic transformations, leading to the accumulation of metabolites that may have important physiological effects on the host as well as on other microbes.
In 2017, I started my PhD at the group of Marnix Medema at Wageningen University to develop a tool to facilitate the genome mining of the gut microbiome for specialized primary metabolism: gutSMASH. This was not going to be a duo journey. We were fortunate to have Michael Fishbach, an expert chemist on small molecules characterization from Stanford University, on board from day one. Having the team assembled, we started by creating a Metabolic Gene Cluster (MGC) collection by iterative homology searches of already characterized pathways. From this preliminary analysis, we got a sense of how the metabolic landscape of anaerobes looked like, which kind of detection rules (and how flexible) were needed and pinpointed the key enzymes and their corresponding domains (also referred in the article as core enzymatic domains). We designed the tool based on the antiSMASH framework: we created detection rules for each pathway using a combination of existing profile Hidden Markov Models (pHMMs) for protein families and customized pHMMs for protein subfamilies of interest, thus making the rules more targeted and robust. At the same time, when analyzing the MGC collection, we found interesting gene cluster architectures having in common the presence of at least one of the core domains but no associated function (some of them were described here in a separate paper). As these could represent a new source of new pathways and/or metabolites we created another set of putative detection rules to be able to capture these interesting MGCs as well. Just in time, Dylan Dodd, also from Stanford University, joined the team with his dilated knowledge on anaerobic metabolism and helped to complete and validate the set of detection rules.
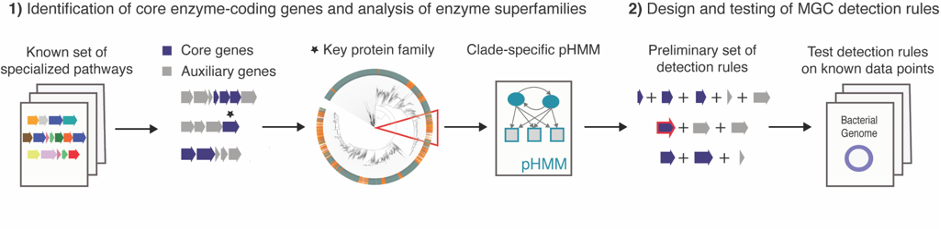
Based on previous experiences, we already knew that validating the performance of such predictions is a challenging task. It implies first of all a deep understanding of the metabolic capabilities of microbes and second, the capacity to verify the presence/absence of these pathways in a wide range of taxonomically diverse bacteria. Being realistic, we performed multiple analyses to validate our predictions: (1) we ran gutSMASH on a set of reference genomes and manually analyzed the MGCs predicted by each rule individually. (2) We checked if we could predict a given pathway by using as input the genome retrieved by a paperBLAST search using the sequence of the key protein as a query and (3) we ran gutSMASH on a handful of metabolically characterized and well-known genomes, and checked whether the reported pathways were successfully predicted (and the other way around).
Once the detection rules were created and validated, the tool was also tailored to meet the needs of the new predictions. That implied creating new databases, new gene functional annotations and code refactoring. Luckily, for any technical questions we could reach Simon Shaw and Kai Blin (current antiSMASH maintainers) from the Technical University of Denmark. In parallel, we decided to explore the metabolic capacities of the most representative genera inhabiting the human gut. Thus, we compiled the genomes of two collections the HMP (Human Microbiome Project) and the CGR (Culturable Genome Reference) plus an additional set of Clostridiales to account for their diversity and representation in this ecosystem. Besides presenting gutSMASH, we also wanted to get the full picture in terms of “who does what” in the human gut. Thus, later on in the project, we also included the distribution of several single-protein pathways that are known to contribute to the pool of specialized metabolites circulating in the gut.
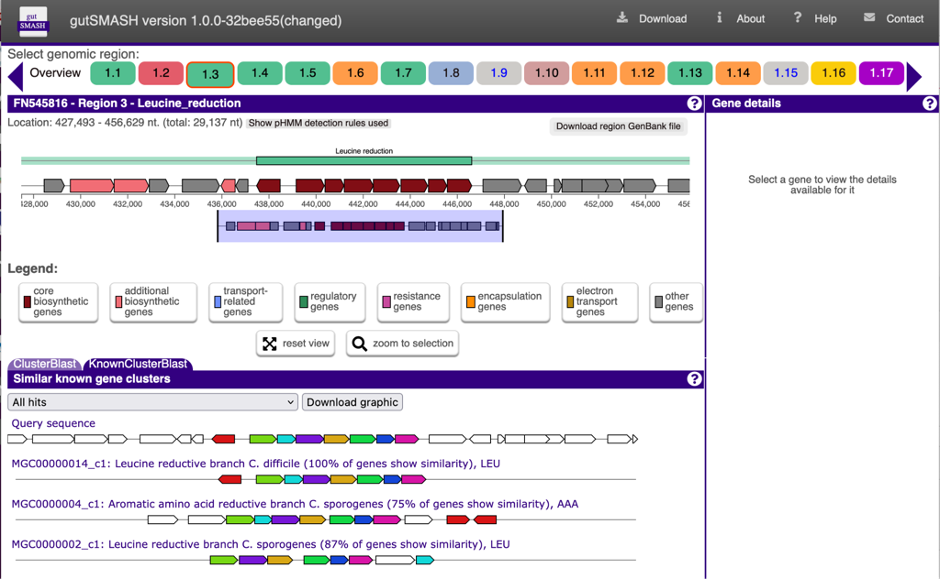
We had now fully developed gutSMASH (now also available via a web server: https://gutsmash.bioinformatics.nl/) and systematically profiled the gut microbiome metabolism, identifying 19,890 gene clusters in 4,240 high-quality microbial genomes. Nevertheless, we believed it would be interesting to assess the representation of these pathways across a human population. For this task, we teamed up with Jingyuan Fu from Groningen University, as we thought the Lifelines Deep (LLD) Cohort samples (metagenomics and metabolomics) were ideal. At this point, Hannah Augustijn, who had co-developed BiG-MAP (an automated pipeline to profile metabolic gene cluster abundance and expression in microbiomes) during her master thesis in our group joined the team. As a member of the group of Jingyuan Fu, she was familiar with both the LLD data and the methods, and was put in charge of computing the prevalence and abundance of each of the gutSMASH-predicted pathways.
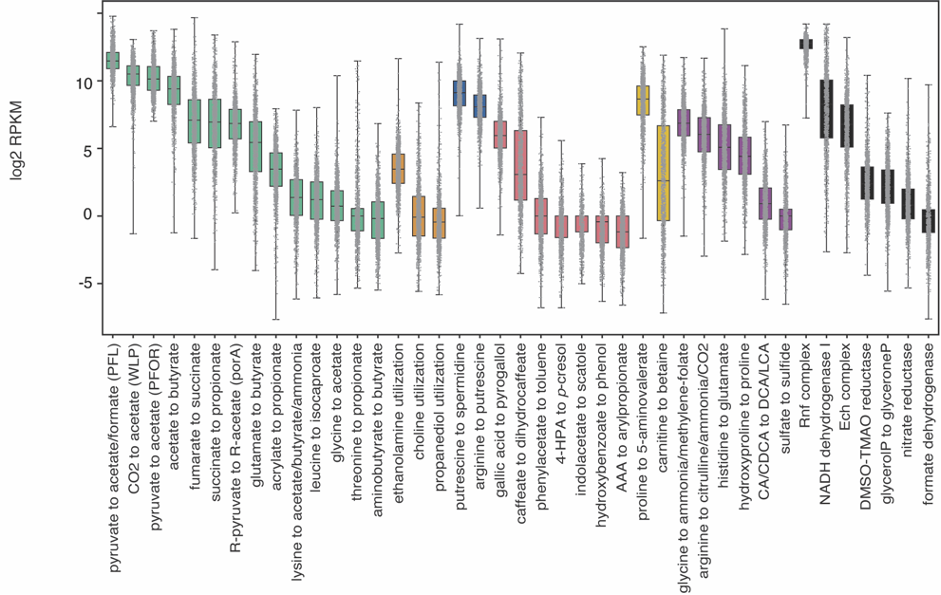
Complementarily, Lianmin Chen, a PhD student with Jingyuan Fu at the time, analyzed the correlation between the abundance of the pathways and the metabolomics data. The results were clear and showed little to no-correlation between the MGC abundance and its end product. We hypothesized that this lack of correlation could be due to complex regulatory processes that affect gene transcription. However, as the metabolites were extracted from plasma, we were not able to rule out the possibility that the results would be different if directly measured from feces. During the revision of the article, another question arose: Despite the fact that the relation between genomic content and the end product is not 1:1, is the pathway expression correlated with metabolite concentrations? To clear this up, we analyzed the iHMP (integrative Human Microbiome Project), which includes paired metagenomic, metatranscriptomic and metabolomic data in the same way. We could find stronger correlations between metatranscriptomic and metabolomic data. Nonetheless, it appeared to vary a lot depending on the pathway. As these results indicate, predicting metabolic outputs from metagenomic or metatranscriptomic data alone may often not be an accurate representation of reality.
I would like to finally highlight the collaborative effort that brought this project to a successful finish after many years of work. I thank all the people who helped us along the journey, and we take the opportunity to make a call to the community to join forces and combine expertise to tackle such complex and multidisciplinary problems as the human microbiome and its role in health.
Source code for gutSMASH is available here.
You can run gutSMASH via its web server here.
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in