Efficiently solving the Schrödinger equation for many molecules at once with deep neural networks
Published in Chemistry

Early in 2019, we came across a preprint from the group of Weinan E proposing a deep neural network-based ansatz function (DeepWF) for variational Monte Carlo solutions of the Schrödinger equation1. We found this approach to be extremely promising. Deep neural networks (DNNs) have led to incredible breakthroughs in many areas of machine learning and modern frameworks such as TensorFlow or PyTorch provide a simple and efficient means of performing gradient descent-based optimization for highly complex models. Combining deep neural networks with quantum Monte Carlo (QMC) thus seemed to be a natural and very promising step forward.
We had not previously worked on QMC methods, but felt that our combined expertise in machine learning for computational chemistry and deep learning-based solvers for partial differential equation could put us in an interesting position to contribute to this emerging field. Our initial goal was to investigate how "deep QMC" could be best applied in a setting where the solution for the Schrödinger equation was not required for a single molecule but a diverse set of different geometries. We knew that many properties such as energies, forces, or properties of excited states, exhibit a high degree of regularity in the space of molecular configurations, meaning that interpolation methods can often accurately predict these properties for new configurations given a sufficient amount of training data. We thus suspected a similar regularity for the wavefunction of the ground state itself, which could be leveraged to drastically improve the optimization of DNN-based models. However, at this point, it was neither clear if this assumption would hold in practice and how a robust algorithm exploiting it would look like.

At the early stages of the project, the interdisciplinary nature of our collaboration and the novelty of deep QMC in general posed a significant challenge. Applied mathematics and computational chemistry are related but still very different fields with their own language and their own way of approaching a task. Finding a sustainable common ground at a time when the success of our general approach was still uncertain and when we were not yet familiar with many of the intricacies of applying DNNs for QMC turned out be quite difficult. In this regard, we believe that stubbornly sticking with the project despite a large amount of uncertainty, regular setbacks and occasional misunderstandings was the key to what would eventually become a successful collaboration.
A highly significant month for the young field of deep QMC was the September of 2019, when the FermiNet2 and PauliNet3 architectures were published almost concurrently on arXiv. Both succeeded in outperforming state-of-the-art ab-initio methods and were the first to demonstrate that DNN-based QMC could indeed be a path towards accurate wavefunctions for previously intractable systems. At a time when we had just ran a first round of explorative toy experiments, the impressive works of both groups left a strong impact on us. They significantly raised the bar on what would be expected from our own work in terms of novelty and accuracy. Furthermore, it was a huge motivation boost for us to see that the initial idea that DNNs could drastically decrease the order of complexity when optimizing high-accuracy QMC models was more than an empty promise and that some very bright people were putting a lot of effort into making it work.
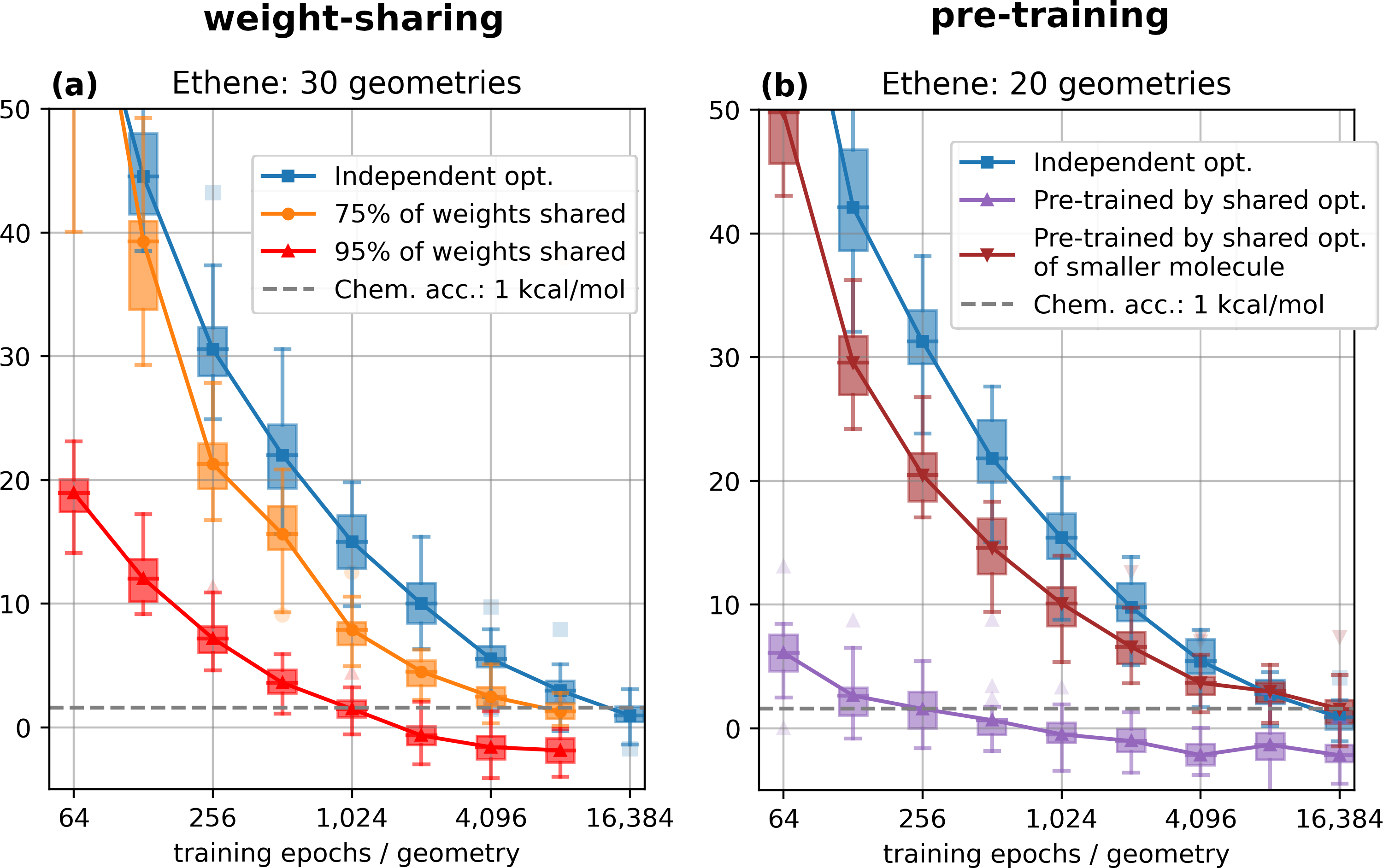
The work we ended up publishing really began to take shape in 2020, when Michael and Leon joined the group of Philipp Grohs as PhD students. Their work drive and impressive skill level turned out to be pivotal for the success of this project. After floating around different ideas and playing with different DNN-based ansatz models and optimization schemes, we came around to make two major decisions regarding the direction this work. First, we settled on weight-sharing as our main approach for realizing possible synergies when optimizing wavefunctions for different configurations at the same time. Weight-sharing had already shown promising results in early numerical experiments and we believed this approach to be methodologically sound. Furthermore, we decided to conduct our investigation with a DNN-based model that was closely following the design of PauliNet. At least for smaller systems, PauliNet achieved state-of-the-art accuracies after relatively few optimization steps, which we believed to be essential to keep the computational cost at bay for a project that would require extensive numerical experiments for sets of up to a hundred different molecular configurations.
Trusting in this setup, we soon realized that weight-sharing does indeed yield consistent and reliable improvements for the optimization of sets of configurations for different molecules. What we did not expect was that weight-sharing worked best when sharing almost all (~ 95 %) of the weights in our model. Our original intuition was that forcing 95 % of the model to perform the same computation across different configurations would make it impossible to find a solution that yields high accuricies for all configurations. We think that this suggests even a stronger regularity of wavefunctions within the space of nuclear geometries than originally anticipated. Further evidence for this is also provided by a very interesting concurrent approach named PESNet4, which was released shortly after our paper was first available as a preprint.
Seeing our results published after such a long journey was really a gratifying experience. Beyond that, we are very curious about which future directions the still young but vibrant field of deep QMC will take. Given the proven potential of the method and the large amount of talent currently involved in this endeavour, we are certain that exciting years are ahead.
References
1. Han, J., Zhang, L., & Weinan, E. (2019). Solving many-electron Schrödinger equation using deep neural networks. Journal of Computational Physics, 399, 108929.
2. Pfau, D., Spencer, J. S., Matthews, A. G., & Foulkes, W. M. C. (2020). Ab initio solution of the many-electron Schrödinger equation with deep neural networks. Physical Review Research, 2(3), 033429.
3. Hermann, J., Schätzle, Z., & Noé, F. (2020). Deep-neural-network solution of the electronic Schrödinger equation. Nature Chemistry, 12(10), 891-897.
4. Gao, N., & Günnemann, S. (2021). Ab-Initio Potential Energy Surfaces by Pairing GNNs with Neural Wave Functions. arXiv preprint arXiv:2110.05064.
Follow the Topic
-
Nature Computational Science
A multidisciplinary journal that focuses on the development and use of computational techniques and mathematical models, as well as their application to address complex problems across a range of scientific disciplines.

Related Collections
With Collections, you can get published faster and increase your visibility.
Next-Generation Digital Twins for Drug Discovery
Publishing Model: Hybrid
Deadline: Mar 22, 2027



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in