Extracting mechanistic details from biomolecular simulations: The case of Watson-Crick to Hoogsteen DNA base pairing transition pathways
Published in Protocols & Methods
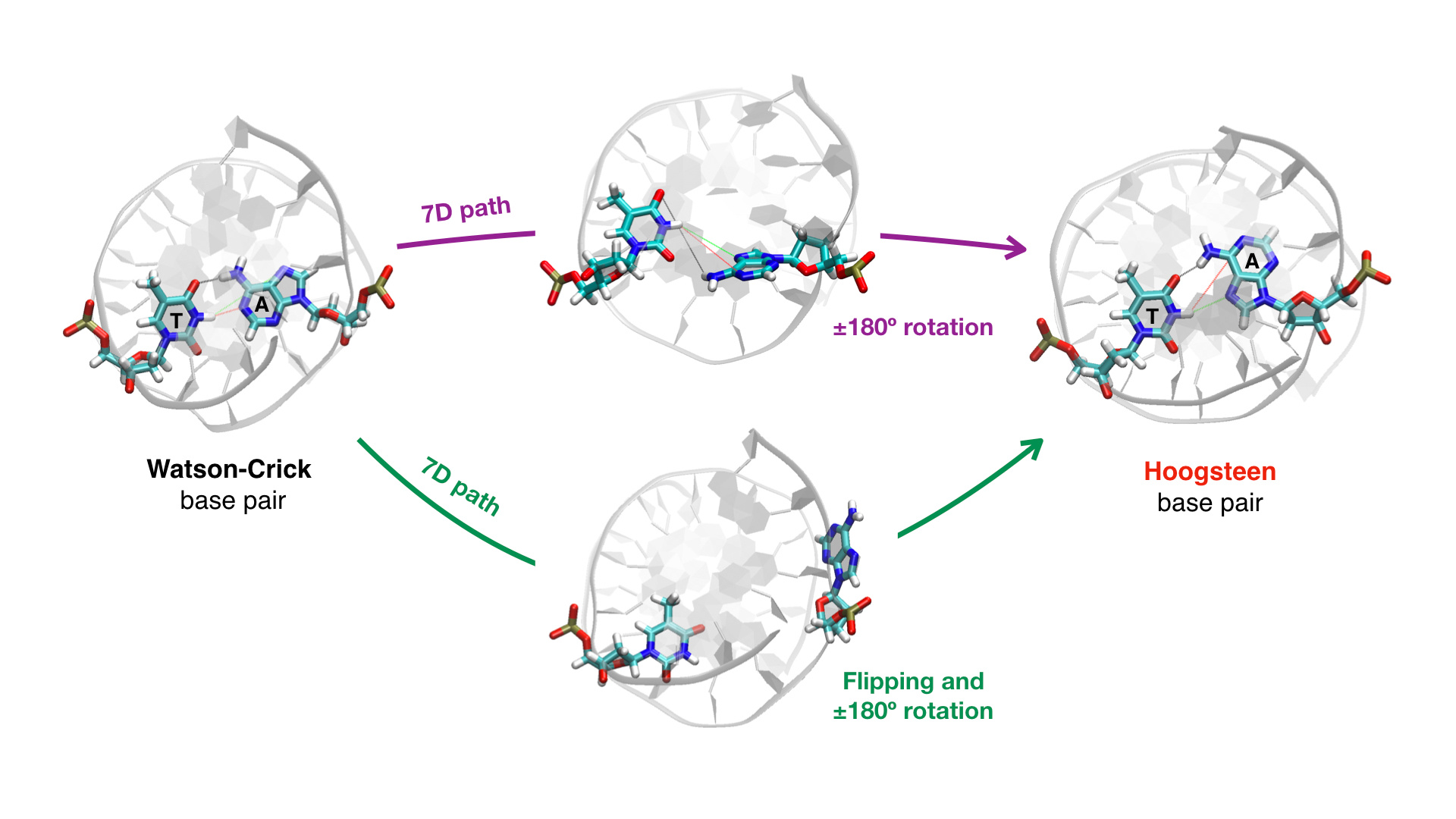
In 1959—six years after Watson and Crick introduced their famous DNA structure—Karst Hoogsteen published an alternative conformation in which nucleobases can pair with each other. The now called Hoogsteen base pairing is characterized by a 180º rotation of the purine base around the glycosidic bond with respect to the Watson-Crick configuration, resulting in an "upside-down" adenine or guanine. In the last decade, the Hoogsteen motif has caught attention because of its newly discovered role for a number of biological functions and its transient presence in duplex DNA. Several experimental and computational efforts have been deployed in order to understand the transition mechanisms that connect the Watson-Crick and the Hoogsteen states. Because the transitions occur quickly and rarely, experimental observation is challenging and molecular dynamics (MD) simulations require special sampling techniques. Conjugate peak refinement and umbrella sampling have been used to render free energy landscapes of this process. The surfaces were projected onto two key molecular transition descriptors, or collective variables (CVs), related to the purine base: the rotation around the glycosidic bond, and the flipping outside of the double-helix, mainly toward the major groove. Two kinds of pathways have been identified, inside and outside, differentiated by the absence or presence of flipping:
But there is still much to be discovered about the mechanistic details of this transition.
In our book chapter, we demonstrate the power of our in-house developed adaptive path collective variable by investigating the Watson-Crick to Hoogsteen transition considering many more CVs than previous studies (seven in total!). While standard methods suffer from an exponential increase in computational cost with the number of CVs, the path formalism has a linear—and even sub-linear—performance scaling; thus allowing the study of highly complex transitions. The approach is based in the following idea: rather than sampling and building a free energy surface on the high-dimensional CV-space explicitly, we explore a one-dimensional progress parameter along a flexible curve that connects two known stable states in CV-space. By iteratively optimizing the curve to follow the average sampled density, an indicator of the underlying free energy gradient, we can converge to the average transition path, or the minimum free energy path, which provides key mechanistic details about the transition. Moreover, by applying an enhanced sampling method (e.g., metadynamics) we can obtain a free energy profile along the path. Our path-metadynamics framework has successfully been applied in various systems and levels of theory. Here you can see it in action, capturing the C7eq to C7ax transition for alanine dipeptide in vacuo:
In the protocol chapter we show, step by step:
- The theory behind the adaptive path CV and how to perform enhanced sampling on it.
- The computational implementation of the adaptive path CV in the PLUMED plug-in, which can be used to perform enhanced sampling with multiple MD packages.
- The simulation setup for the DNA system.
- The definition of seven CVs for the base pairing transition, based on key structural signatures of the two motifs; and the localization of the two stable states in the seven-dimensional CV-space.
- How to test the CV set by performing steered MD.
- How to perform, analyze convergence and interpret results from a multiple-walker path-metadynamics simulation.
- How to optimize an adaptive path based on trajectory data from transition path sampling; and how to get a free energy profile using umbrella sampling along an already optimized path.
- What can we learn about the transition mechanisms and the molecular system at hand.
- And many more tips and details...
We hope you find this chapter useful when setting up your own adaptive path and enhanced sampling simulations. Feel free to contact us if there are any questions. From our part, we are developing further extensions and improvements for the method; as well as continuing our research on finer details of the Watson-Crick to Hoogsteen transition, and other biomolecular systems.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in