Finding microbes that produce diverse chemistry
Published in Microbiology
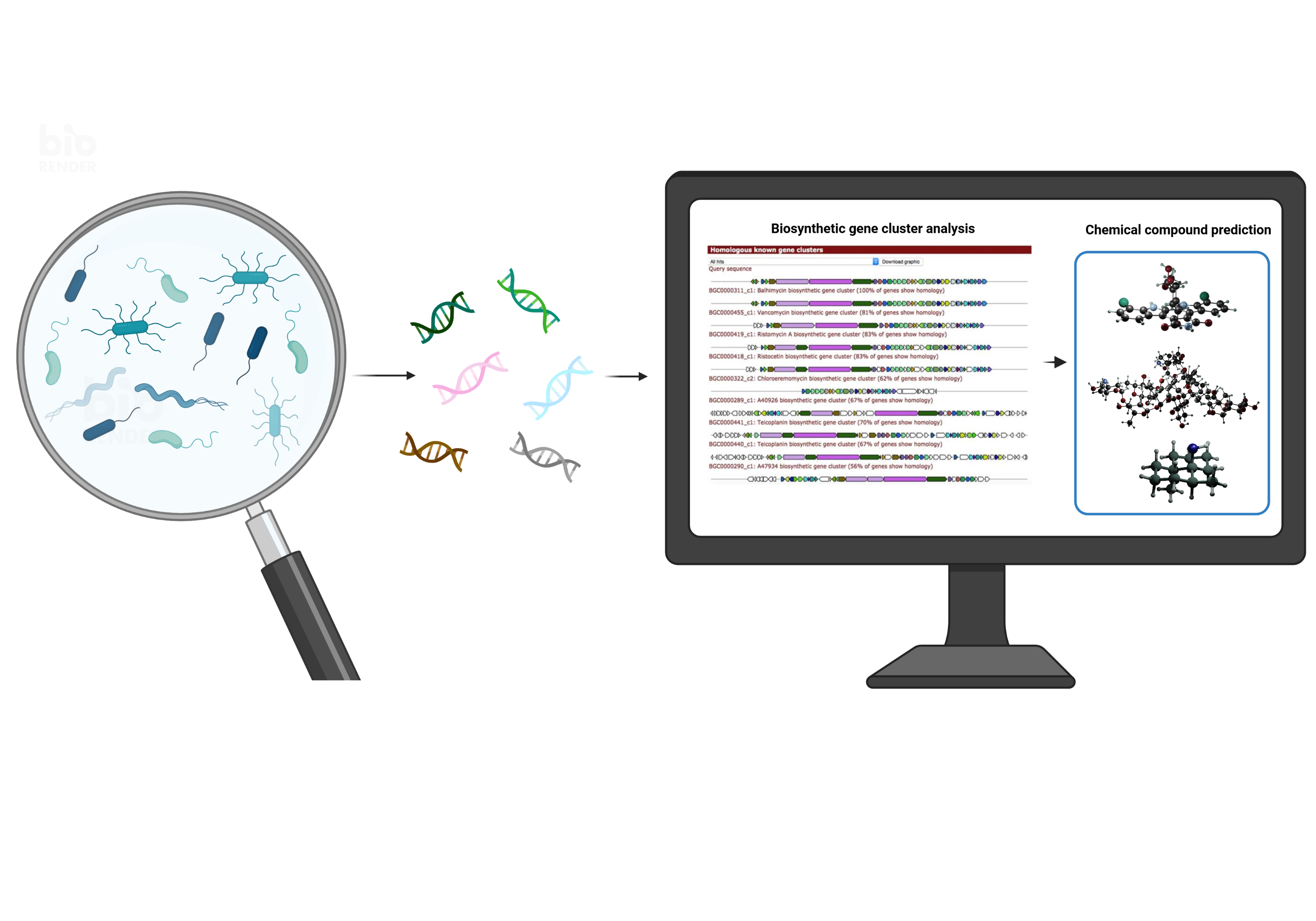

Bacteria produce a vast number of specialized metabolites, also known as secondary metabolites, which are used as defensive agents, for nutrient acquisition and signaling. As Natural Products (NP), these molecules are particularly important as the basis for novel drugs because of their bioactivity as antibiotics, anti-cancer, and immunomodulatory agents.
Enjoying natural product conferences in the past couple of years, we always noted that researchers looking for novel compounds followed, or proposed to follow, different strategies. Some of them pursued the search for novel compounds in unusual environments of hard-to-reach areas. The hope was to identify the urgently needed, novel antimicrobials from new strains of well-known antibiotic-producing taxa such as Streptomyces or related actinomycete genera. Other researchers pointed out the over-mining of those well-studied bacteria and the high rates of rediscovery of known compounds, pinpointing so far overlooked bacteria as sources of more unexplored chemistry. Interestingly, most arguments were solely based on experience or anecdotal evidence from small-scale analyses of certain taxa.
As natural product researchers with strong interests in computational analysis and genome mining, we started to wonder whether analysis of large datasets could be used to identify the best mining strategies. The production of bacterial secondary metabolites is in fact encoded by so called biosynthetic gene clusters (BGCs), and bacterial genomes are plenty at hand. Moreover, recently established databases such as MIBiG (https://mibig.secondarymetabolites.org/) and NP Atlas (https://www.npatlas.org/) (for experimentally characterized biosynthetic gene clusters and chemical compounds, respectively) provide the necessary training data. Tools such as antiSMASH (https://antismash.secondarymetabolites.org/#!/start) can detect BGCs within bacterial genomes faster and more accurately than ever, and the recently developed BiG-SLiCE tool (https://doi.org/10.1093/gigascience/giaa154) is able to group similar gene clusters into families that encode for the biosynthesis of similar or identical compounds. Nonetheless, we noticed that some challenges remained. First of all, bacterial taxonomy: whoever has worked with bacterial genome data before must have noticed that names do not necessarily reflect the evolutionary relationships. What has been called a Streptomyces before, might not be a Streptomyces anymore. Additionally, there is no unambiguous definition of the different taxonomic groups, meaning that some of the genera are very narrowly defined, whereas others comprise genetic distances equivalent to those found across four or five other genera. So, we would have compared single houses with entire towns when comparing biodiversity according to the current nomenclature, which did not seem fair. To overcome this challenge, we defined so-called REDgroups as groups of equal relative evolutionary distance based on the genome-based GTDB taxonomy (https://gtdb.ecogenomic.org/). Doing so, we noticed that, for example, Streptomyces was subdivided into 21 different REDgroups, which could explain why researchers found such a broad chemical diversity within this genus. It turned out that, even among these 21 groups, many contain some of the most diverse repertoires of gene cluster families.
But Streptomyces species have obviously been sequenced a lot, leading us to the other challenge we had to overcome: the sequencing bias. For the sole fact that pathogenic bacterial strains or biotechnologically relevant strains have been sampled and sequenced extensively, more chemical diversity was likely to be found in these species than in the under-sequenced ones. We could not entirely solve this problem. But using rarefaction curves and biodiversity extrapolation methods verified by random sampling, enabled us to quantify the potential chemical diversity encoded in bacteria and to draw conclusions about sampling strategies.
In our analysis of large datasets, we see that both compound hunting strategies described above are valid. Overall, by calibrating the granularity of the grouping of gene clusters to natural product classes in the NP Atlas (https://www.npatlas.org/), we were able to estimate that only 3% of natural product classes found in sequenced taxa have been experimentally characterized. Streptomyces and related actinomycetes encode by far the most chemical diversity and there is much more chemistry left to discover within specific REDgroups. Furthermore, it seems that the majority of bacteria do not encode the biosynthesis of notable amounts of known natural product classes. Within the bacterial kingdom there are randomly distributed genera with high genomic potential to produce specialized metabolites, which have not been subjected to any chemical analysis. Not yet.
Additionally, with both metagenomics-derived gene clusters and compounds becoming more accessible, we were curious to examine how large their contribution is to the chemical diversity of the bacterial kingdom. Our results indicate that a great deal of unique diversity lies in metagenome-assembled genomes, that is, in currently unculturable bacteria. This observation, combined with the fact that the cultured bacterial strains comprise the minority of nature’s taxonomic diversity, proves once again the future potential of metagenomics. Furthermore, bioprospecting efforts would also benefit from an investigation of the possible connections between unique chemical diversity and biogeography, an undertaking that was only partially possible for us to accomplish due to a lack of suitable and consistent metadata. As the field of metagenomics evolves at high speed, we are confident that the documentation process will soon be standardized and more analyses will be possible. Nonetheless, we were already able to demonstrate that the inclusion of metagenome-assembled genomes in natural product discovery efforts can increase the estimated chemical potential of the bacterial kingdom by at least 20%.
To recap, our project was inspired by the concept that there is so much genomic data publicly available that we should make use of to generate a global overview of chemical diversity in the bacterial kingdom and transform compound discovery into a “data-driven” process. We ended up with two encouraging notions: first, there is a lot of material to learn from and we should not hesitate to make use of it; second, we should not be afraid to run out of novel bacterial sources of diverse chemistry. The challenge left is to identify the compounds.
All our findings are published here: https://www.nature.com/articles/s41564-022-01110-2
Cover art was created with BioRender.com by Libera Lo Presti
Follow the Topic
-
Nature Microbiology
An online-only monthly journal interested in all aspects of microorganisms, be it their evolution, physiology and cell biology; their interactions with each other, with a host or with an environment; or their societal significance.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in