Fresh DHFR complex from a fresh crystal
Published in Chemistry
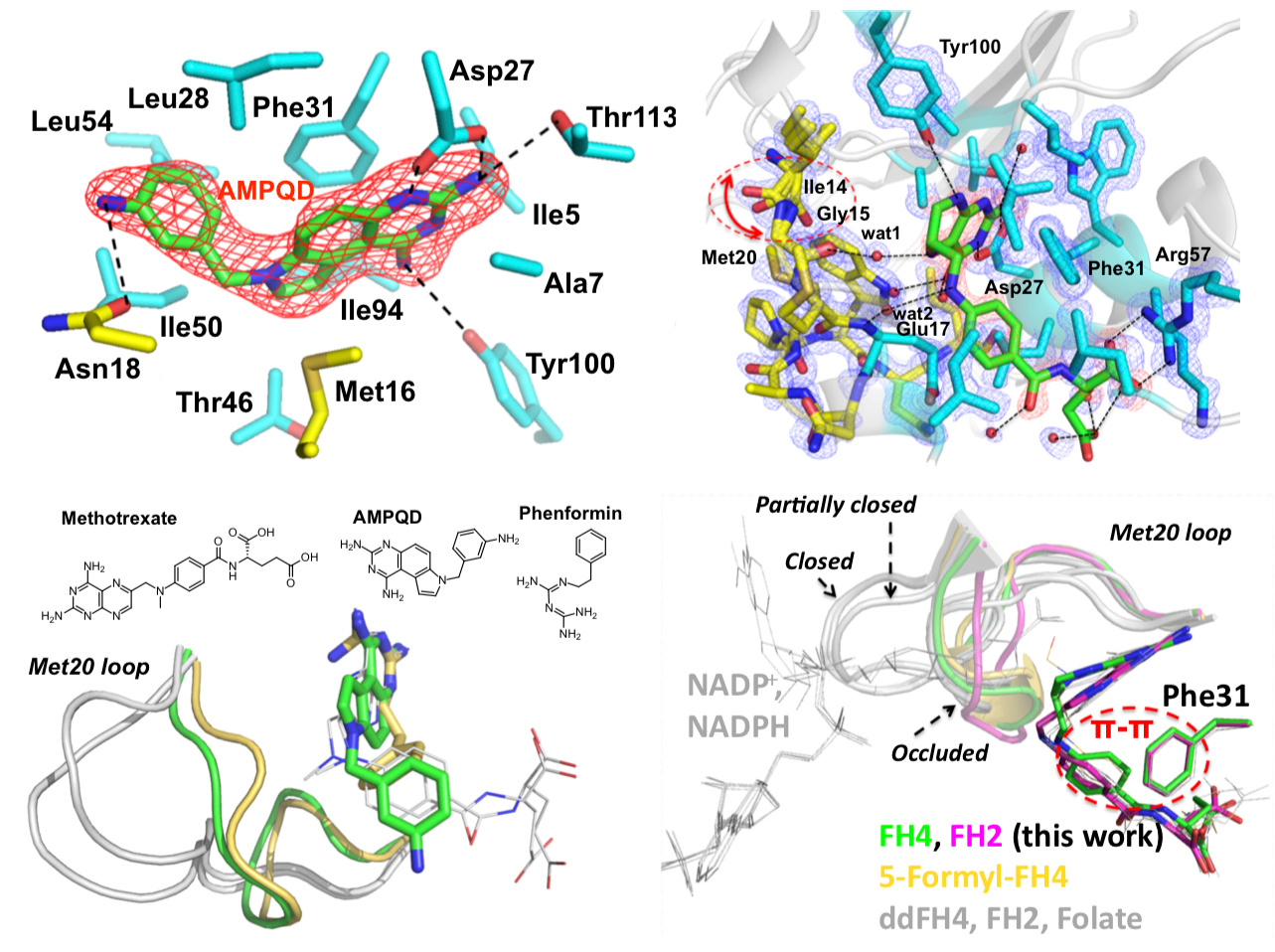

The enzyme dihydrofolate reductase (DHFR) was discovered in the 1950s and is of interest because its catalytic product tetrahydrofolate acts as a one-carbon carrier and plays an essential role as a building block of DNA bases. Interestingly, a few years before the discovery of the enzyme, the clinical effectiveness of its inhibitor aminopterin was demonstrated to treat acute leukemia in children, resulting in temporary remissions in certain patients. This pointed out the possible clinical importance of DHFR inhibitors, an observation that is still true today. Indeed, DHFR is a target for chemotherapy and antibiotics. While the sequence identity of Escherichia coli and human DHFRs is 30%, they have similar overall structures and catalytic properties including the fact that product release is the rate-limiting step. Despite these similarities, their conformational dynamics are quite different. This is partially attributable to the differences in the structural properties of the active site loop. For example, in all experimental structures of human DHFR, the active site loop adopts so-called “closed” conformations that readily accommodate the entry of the nicotinamide moiety of the cofactor NADPH to perform oxidoreduction catalysis. In contrast, distinct “closed” and “occluded” conformations of E. coli DHFR have been captured by X-ray crystallography, NMR, and single-molecule kinetics. Crucially, the “occluded” conformations disallow the entry of nicotinamide to reduce the dihydrofolate substrate, and unlike in human, E. coli DHFR cycles between closed and occluded conformations during catalysis.
The motivation of our crystal structural work in the well-established DHFR field resulted from a computationally driven structure-based drug discovery effort. Our original goal was to identify the binding pose of a slow-onset nanomolar inhibitor of DHFR, AMPQD that was predicted using the FINDSITEcomb virtual ligand screening algorithm. This led us to solve the structure of the inhibitor complex (PDB ID: 6CQA) which happened to be caught in the occluded conformation that contrasts with the documented methotrexate complexes which were found in the closed conformations. Furthermore, we accidentally discovered that the time course of decay of its co-purified native product tetrahydrofolate in enzyme crystals takes on the order of a couple of days and is associated with structural changes that can be directly followed by X-ray crystallography (see below).
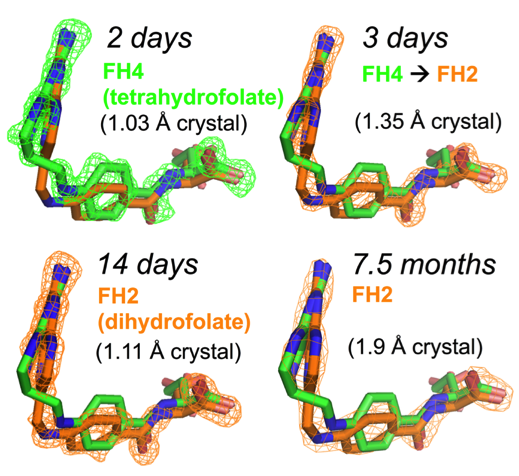
Our solved tetrahydrofolate-bound E. coli DHFR structure (PDB ID: 6CW7) in the occluded conformation is an important piece in the big picture of the catalytic cycle and mechanism of DHFR; this represents the first time that the crystal structure of single domain DHFR was solved with its tetrahydrofolate product. To obtain the hitherto unsolved bound tetrahydrofolate structure, we crystallized the as-purified DHFR without any dialysis or other pretreatment. This points out that the timing of crystal harvesting can matter in terms of not only structural quality but also the structures that are determined. Our crystal structures provide the structural basis for the slow product release and corroborates previous NMR and computational studies which suggested the persistence of the physical properties of the transition state conformation of the enzyme reaction into the product bound state.
Our research has now been published in Communications Biology.
https://www.nature.com/articles/s42003-018-0236-y
Authors: Hongnan Cao, Mu Gao, Hongyi Zhou and Jeffrey Skolnick
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026
DNA repair and human disease
Publishing Model: Hybrid
Deadline: Oct 31, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in