From hundreds to thousands of genes: new advances in identifying small proteins in a minimal cell
Published in Microbiology, Protocols & Methods, and Cell & Molecular Biology
The function and structure of a living organism are determined by its proteome, which consists of its proteins. Predicting a proteome is possible through the genetic information stored in an organism's genome. This process starts with DNA, where genes are encoded in nucleotide sequences. Genes are transcribed into messenger RNAs (mRNAs), which act as templates for protein synthesis during translation, combining amino acids. Advances in sequencing technologies have transformed our ability to study whole genomes and decipher their genetic information, allowing researchers to identify protein-coding genes, predict their functions, and uncover genetic compositions relevant to traits or diseases.
The identification of gene sequences encoding proteins within a whole genome relies on the universal genetic code, where specific 3-nucleotide combinations, namely codons, encode protein starts and stops (e.g., AUG and UAG, respectively). Identifying these initiation and stop positions delineates protein Open Reading Frames (ORFs) corresponding to genes. Using simple probabilistic models, we can demonstrate that an ORF will likely encode a protein when it exceeds 100 amino acids in length. This assumption forms the basis of the initial step in every genome annotation process, where proteins are mapped. However, one might inquire: what about proteins shorter than 100 amino acids? The answer: we typically disregard small ORFs (smORFs) to mitigate false positive gene calls as smORFs are significantly more prevalent than regular ORFs, around 40 smORFs for each ORF, but more frequently not translated into proteins (Figure 1). Therefore, distinguishing whether a smORF will encode a small protein is far from straightforward.
Nevertheless, although challenging to validate, smORFs can encode proteins. Initially identified serendipitously in screening assays, smORF-encoded proteins (SEPs) represent a large pool of little-understood molecules with diverse and relevant functions in cells. For example, in bacteria, SEPs play essential roles in processes such as sporulation, influx inhibition, photosynthesis, cell division, stress sensing, and antibiotic resistance. Additionally, secreted SEPs can contribute to communication and competition in microbial communities, synchronizing cellular reactions between microbes or acting as antimicrobial peptides to eliminate other individuals.
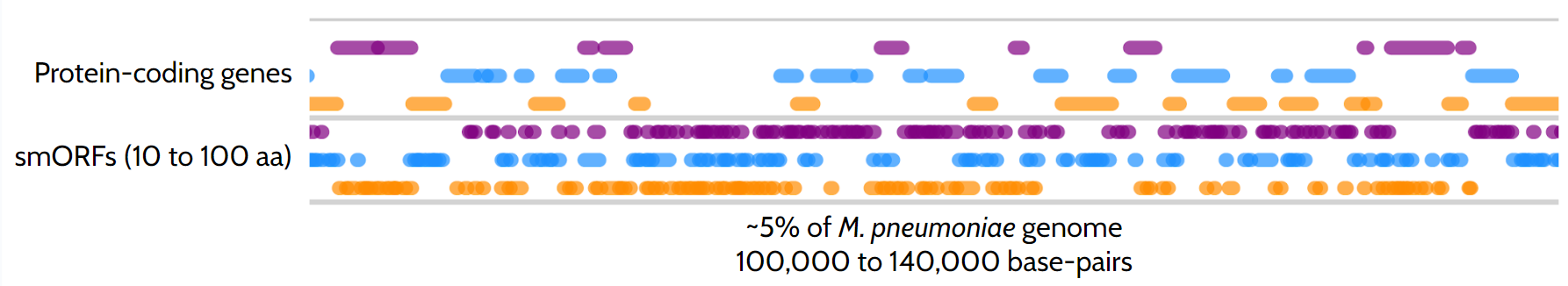
While specific bioinformatic tools have been developed to predict which smORFs translate to SEPs (increasing the number of genes by up to 40% depending on the study and organism), these proteins are often overlooked in genomic analysis due to a lack of experimental evidence. Advances in methods like mass spectrometry and ribosome profiling have enabled high-throughput analysis of proteomes. However, mass spectrometry relies on digesting proteins into unique smaller fragments, which makes it challenging to identify smaller proteins with certainty. Ribosome profiling, while useful, can be noisy and may struggle to identify SEPs translated from smORFs that overlap with larger ones.
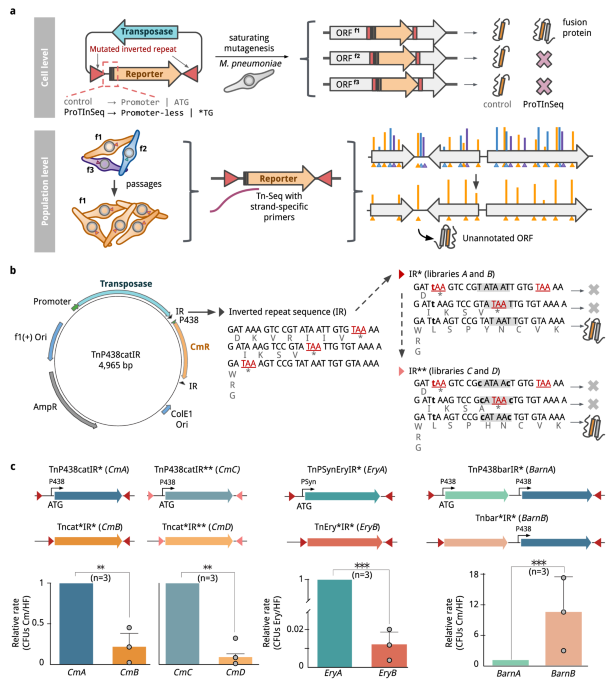
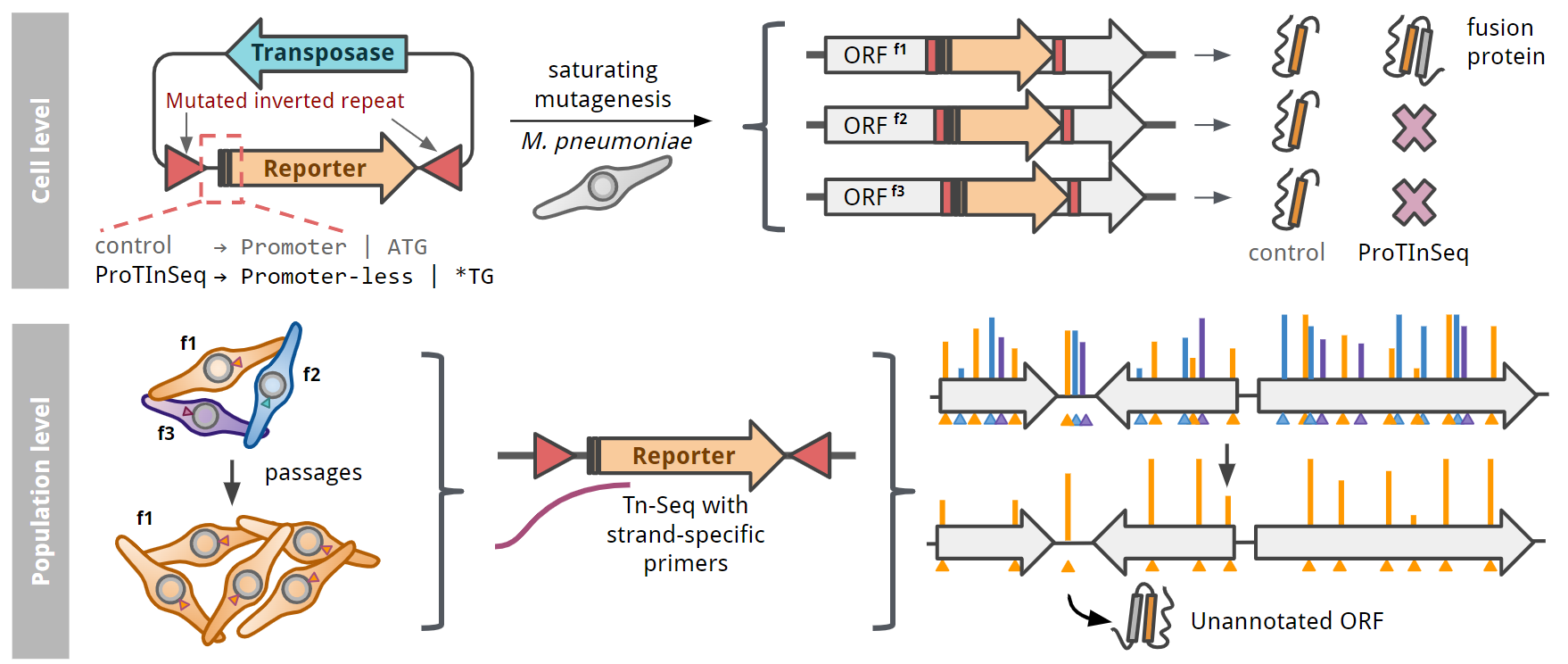
Here we introduce ProTInSeq, a method that uses random transposon mutagenesis and sequencing (Tn-Seq) to explore a proteome of interest. In a regular Tn-Seq experiment, a pool of cells undergoes a process where specific positions in the genome are randomly disrupted. Living cells are then selected based on their growth, as those with an affected essential gene will disappear from the population. Notably, a gene's essentiality in an organism can change depending on various factors, such as genetics or the environment, like during an infection. In ProTInSeq, we use special transposons carrying mutated genes that are only expressed when inserted in-frame to a protein-coding ORF (Figure 2).
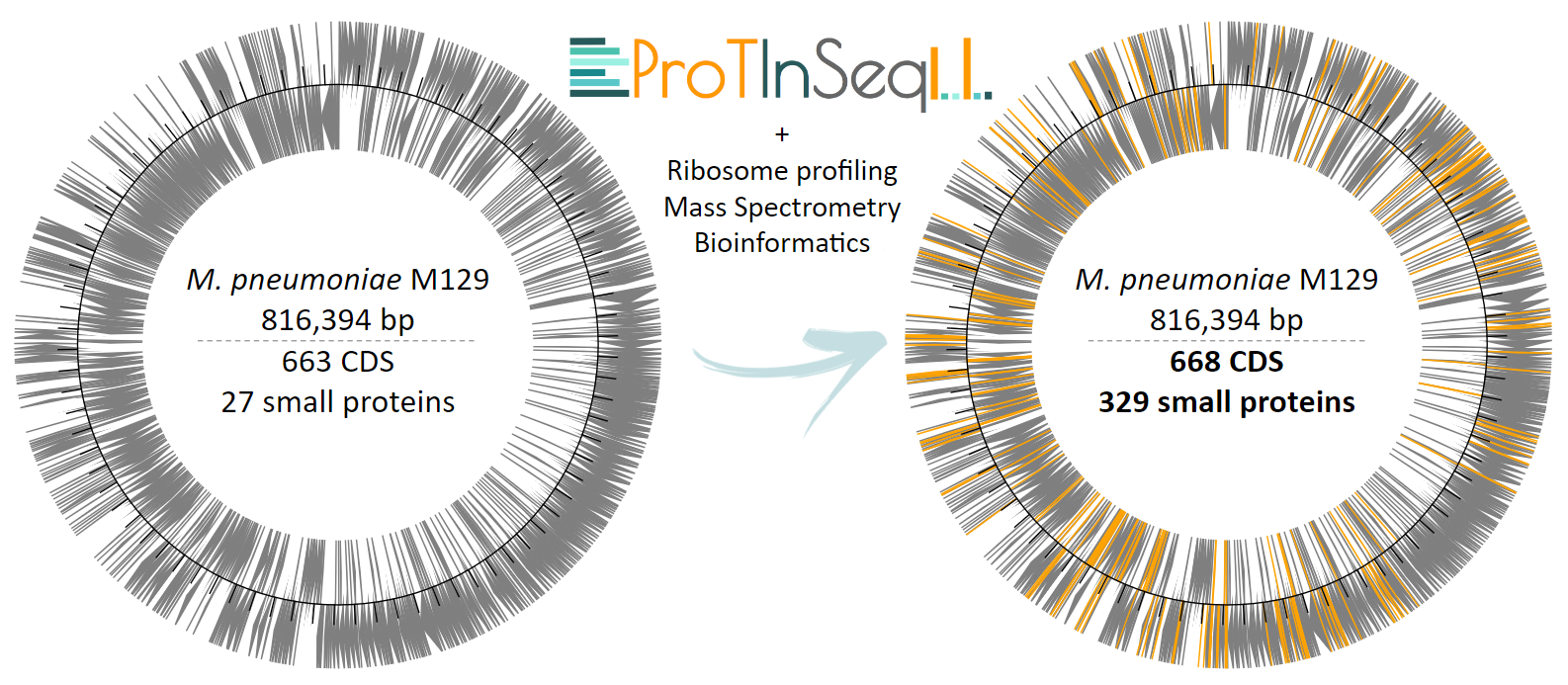
In the minimal genome of the bacterium Mycoplasma pneumoniae, ProTInSeq identifies 80% of known proteins, along with 153 small proteins (SEPs) and 5 proteins larger than 100 amino acids that other methods missed. We also confirmed the reliability of our method by selecting examples of SEPs expressed from regions previously thought to be non-coding, as well as those overlapping with larger genes. Our method also revealed a higher number of SEPs with predicted functions (32%) compared to other experimental approaches, indicating its effectiveness in identifying valuable targets for further investigation. Notably, many SEPs show antimicrobial properties, suggesting their potential in addressing antibiotic resistance. Furthermore, ProTInSeq allows us to measure protein levels, uncover details about their stability and half-life, and identify the topology of proteins in the cell membrane.
When we integrate our method with other computational and experimental techniques, we observe that the genome of M. pneumoniae, previously thought to encode 690 proteins, actually encodes 997 proteins, which comprises a 43% increase in the number of coding genes (Figure 3). To put this in perspective, Escherichia coli, a commonly used bacterial model, has approximately 4,000 proteins in its proteome, while a human's proteome is predicted to contain around 20,000 proteins. If the trends observed in M. pneumoniae extend to these organisms, the number of SEPs awaiting annotation in their proteomes could be in the thousands. Overall, future research on SEPs holds the potential to significantly enhance the functional capabilities of organisms and address challenges such as antibiotic resistance, while also fostering biotechnological innovation.
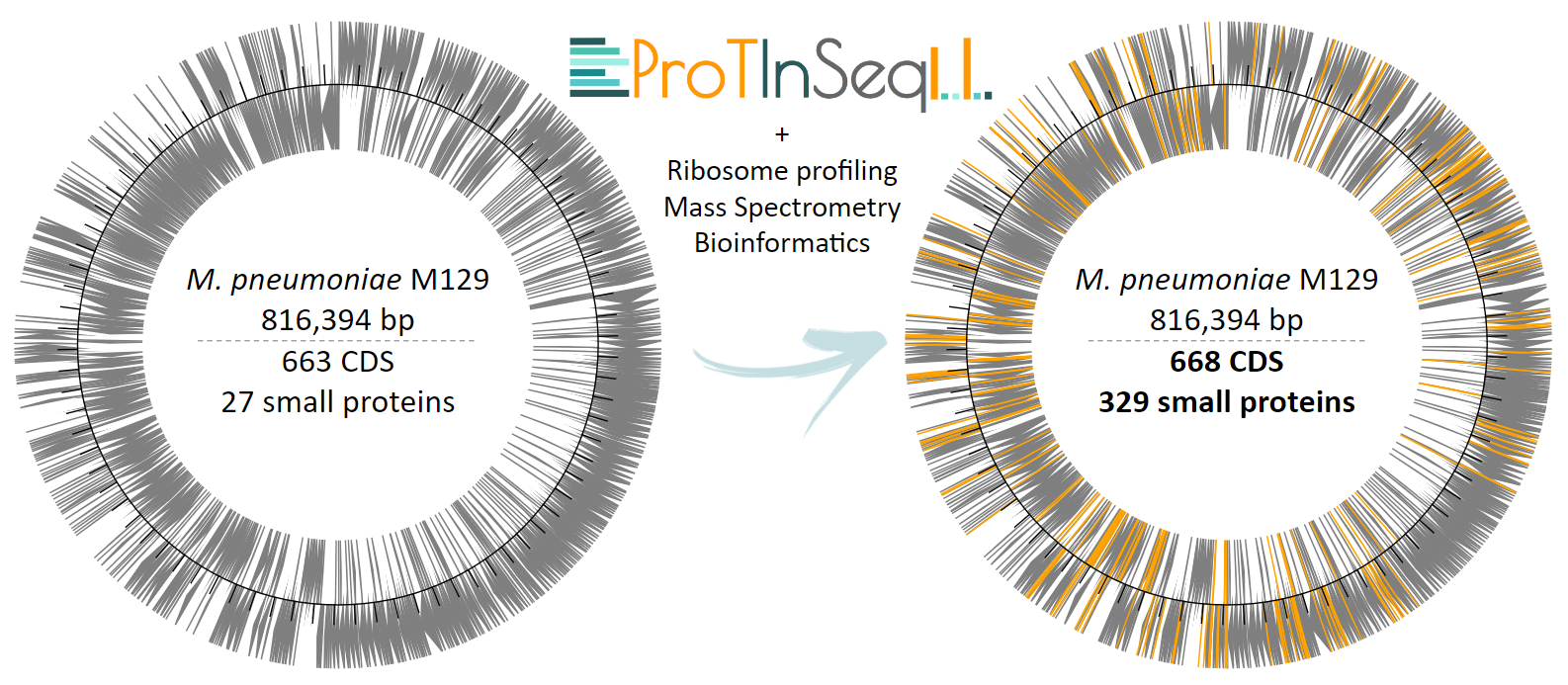
Figure 3. Summary of the increase in both the number and location of genes within the genome of M. pneumoniae following the integration of ProTInSeq-identified SEPs, along with complementary experimental techniques and bioinformatics.
In summary, ProTInSeq represents a flexible, cost-effective alternative experimental method for exploring SEPs through DNA sequencing. This technique can be implemented in other living systems, providing a quantitative tool to characterize SEPs and the structural and physical parameters of a proteome of interest. We envision ProTInSeq as a tool to guide the discovery of novel protein sequences, serving as the first step in prioritizing SEPs for further functional characterization by applying this method in different experimental conditions. In an era where available genome sequences grow exponentially, and considering the pivotal roles of SEPs, we believe ProTInSeq can lead to the discovery of novel SEPs with a direct impact on essential microbial processes and the health of animals and plants.
Read more about this in our recent publication entitled ProTInSeq: transposon insertion tracking by ultra-deep DNA sequencing to identify translated large and small ORFs | Nature Communications.
I am a computational biologist with expertise in microbiology and artificial intelligence-based bioinformatic approaches to understand complex genomic and functional aspects. Specifically, I am interested in studying the prevalence, function and relevance of small proteins in microbes and microbiomes.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Biosensing
Publishing Model: Hybrid
Deadline: Sep 30, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in