Genome-centric investigation of bile acid metabolizing microbiota of dairy cows and associated diet-induced functional implications
Published in Microbiology
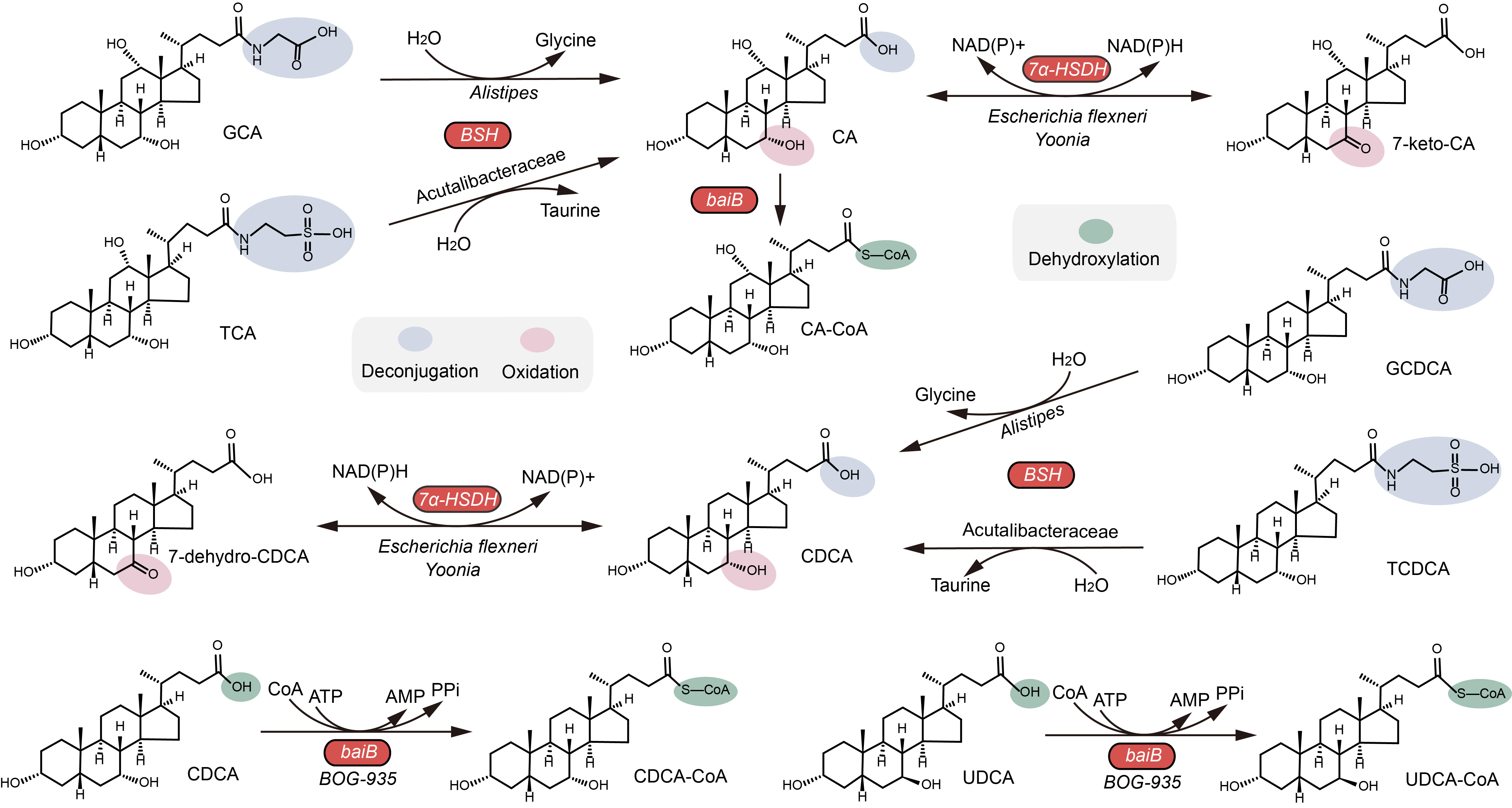

Bile acids (BAs), an important class of biologically active metabolites, are synthesized from cholesterol in the liver. BAs act as important signaling molecules in microbe–host interactions, significantly contributing to numerous physiological processes, including inflammatory responses, lipid metabolism, glucose regulation, and intestinal motility. Gut bacteria are essential players in BA metabolism. To date, the BA transformation pathways of intestinal microorganisms vary but can be classified into four distinct categories: (1) bile salt hydrolases that mediate the deconjugation of conjugated bile salts into deconjugated BAs; (2) dehydroxylation by BA-inducible gene clusters that catalyze a multi-stage process; (3) oxidation; (4) epimerization by dehydrogenase–mediated hydroxysteroids for further modifications of various secondary BAs. Despite the increasing interest in identifying BA-related microbial species and enzymes in monogastric animals, the lack of knowledge of BA metabolism in ruminants limits applications regarding the targeted modulation of microbe–host interactions for improved production and health of livestock such as dairy cows.
Dairy cows are commonly viewed as important microbe–host symbiotic paradigms that can convert low-value plant biomass into highly nutritious milk. With the rapidly rising global population, the demand for dairy products is significantly increasing, resulting in the progressive replacement of the traditional high-forage diet with a high-grain diet. The economic and production benefits associated with a high-grain diet have resulted in its extensive implementation on commercial dairy farms; however, the potential disruption of the cow microbiome associated with this diet is of concern. A high-grain diet can alter physiological homeostasis, including the accumulation of fermentable carbohydrates, rumen pH, and microbial ecology. These microecosystem disorders reportedly contribute to greater susceptibility to metabolic diseases in dairy cows. Despite our awareness of the potential adverse effects associated with the long-term feeding of a grain-based diet on host health, previous studies have primarily focused on assessing common markers, such as volatile fatty acids (VFAs), lipopolysaccharides, lactate, and bioamines in the rumen. Information on BAs linking intestinal microorganisms and hosts during grain intervention in dairy cows is scarce.
In the present study, we used shotgun metagenomics to profile microbial metagenome-assembled genomes (MAGs) and targeted metabolomics to quantify BA pools from content samples of six intestinal regions in Holstein cows (universal dairy cows) fed forage-based versus grain-based diets. Our genome-centric analysis of the BA transformation pathways of 108 intestinal content samples from dairy cows identified 372 high-quality metagenome-assembled genomes (MAGs) that were involved in BA deconjugation, oxidation, and dehydroxylation pathways. Compared with the human and pig gut microbiomes, that of dairy cows had a lower proportion and diversity of microorganisms involved in BA transformation, characterized by the enrichment of host-derived conjugated BAs in the duodenum and jejunum and the prevalence of microbiota-derived unconjugated BAs in the large intestine. Comparative genome analysis showed that bile salt hydrolase (BSH)-carrying microbial populations withstood stressful intestinal conditions via the ability to degrade host mucin, sporulation, and thiamine biosynthesis. The classification of 439 BSH homologs from the intestinal microbiome of dairy cows into 12 clusters based on SSN revealed diverse evolution, taxonomy, signal peptides, and ecological niches for the different clusters. Further integration of microbial genomic signatures, targeted BA pools, and host transcriptomes showed that the strains of Firmicutes bacterium CAG-110 processed the increased abundance of BSHs from Cluster 1, coinciding with changes in the colon concentration of cholic acid, a compound intricately linked to intestinal inflammation via the lectin pathway after grain intervention.
This study is the first to use a genome-centric approach and whole intestine-targeted metabolomics to reveal microbial BA metabolism and its diet-induced functional implications in dairy cows. Our findings provide important clues for precise manipulation of microbial populations to improve animal health.
- Wang D, Doestzada M, Chen L, Andreu-Sánchez S, van den Munckhof ICL, Augustijn HE, et al. Characterization of gut microbial structural variations as determinants of human bile acid metabolism. Cell Host Microbe. 2021;29:1802-1814.e5.
- Sinha SR, Haileselassie Y, Nguyen LP, Tropini C, Wang M, Becker LS, et al. Dysbiosis-induced secondary bile acid deficiency promotes intestinal inflammation. Cell Host Microbe. 2020;27:659-670.e5.
- Guzior DV, Quinn RA. Review: microbial transformations of human bile acids. Microbiome. 2021;9:140.
- Zheng X, Chen T, Jiang R, Zhao A, Wu Q, Kuang J, et al. Hyocholic acid species improve glucose homeostasis through a distinct TGR5 and FXR signaling mechanism. Cell Metab. 2021;33:791-803.e7.
- Eisler MC, Lee MR, Tarlton JF, Martin GB, Beddington J, Dungait JA, et al. Agriculture: steps to sustainable livestock. Nature. 2014;507:32-4.
- McGuffey RK. A 100-Year Review: Metabolic modifiers in dairy cattle nutrition. J Dairy Sci. 2017;100:10113-42.
- Russell JB, Rychlik JL. Factors that alter rumen microbial ecology. Science. 2001;292:1119-22.
- Cai J, Sun L, Gonzalez FJ. Gut microbiota-derived bile acids in intestinal immunity, inflammation, and tumorigenesis. Cell Host Microbe. 2022;30:289-300.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in