Harnessing Type I CRISPR–Cas systems for genome engineering in human cells
Published in Bioengineering & Biotechnology
Since 2012, when Emmanuelle Charpentier, Jennifer Doudna, and colleagues published their landmark paper describing the RNA-guided DNA endonuclease Cas9 (Jinek et. al., Science, 2012 Aug 17;337(6096):816-21), there has been an explosion of interest in both advancing genome engineering tools with Cas9 and mining additional CRISPR-Cas systems. Motivated by the efficiency, facile programmability, and sheer utility of Cas9, researchers have spent the last seven years scouring microbial genomes for alternative CRISPR systems that could offer new biotechnology opportunities. The microbial genome space is vast and only a fraction has thus far been probed. Nonetheless, these efforts have already culminated in the exciting discovery of new RNA-guided DNA targeting systems, such as Cas12a, and even RNA-guided RNA targeting systems, such as those belonging to the Cas13 family. All of this dizzying progress continues to beg two essential questions: (1) what is the extent of the CRISPR system diversity in nature, and (2) how many of these systems can be deployed as tools?
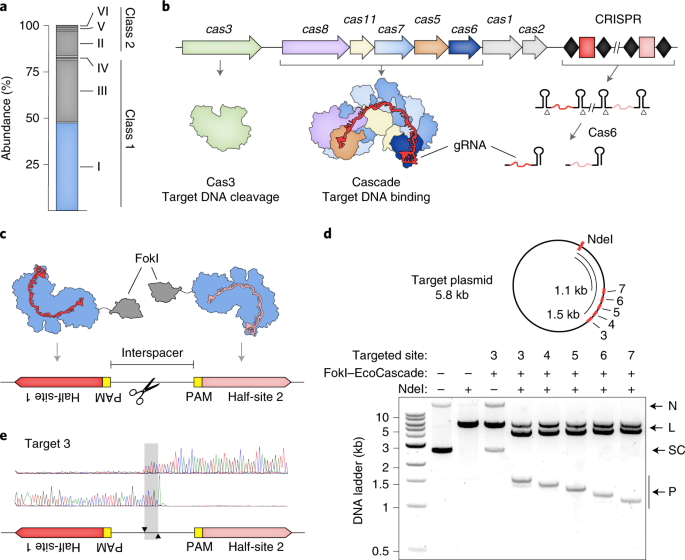
CRISPR systems are divided into two broad classes based on whether they contain single- (Class 2) or multi- (Class 1) protein effector complexes, and then further subdivided into additional types and subtypes depending on the presence of certain “signature” genes and their operon genetic architectures. Thus far, efforts to identify new CRISPR systems for biotechnology applications have largely focused on Class 2 systems, based on the underlying assumption that a single protein effector complex will ultimately be easier to deliver to (or heterologously express in) eukaryotic cells. However, Class 1 systems comprise ~80% of all sequenced CRISPR systems (see Fig. 1). Moreover, Class 1 systems are highly diverse and contain effector complexes that are mechanistically and architecturally distinct from those found in Class 2 systems. For example, Class 1 Type I systems processively degrade their target DNA (if Cas9 is a pair of “molecular scissors,” think of Type I systems as a “molecular paper shredder”), and Class 1 Type III systems target both RNA and DNA and generate signaling molecules in the process.
At Caribou Biosciences, we are using precision genome editing to develop allogeneic immuno-oncology cell therapies and, as part of this mission, we have a longstanding interest in expanding our gene editing capabilities, which includes exploration of new CRISPR-Cas systems. As such, we set out to test whether we could harness multi-protein Type I CRISPR-Cas systems for human genome engineering, thus opening new doors for technology development. The RNA-guided effector complex in Type I-E systems, termed Cascade (CRISPR-associated complex for antiviral defense), contains 11 protein subunits encoded by 5 genes, as well as a 32-nt RNA guide. In the native system, Cascade recruits a helicase-nuclease protein termed Cas3 to initiate the target DNA degradation. In order to avoid the complexity associated with measuring that activity in a human genome, we decided to fuse the dimerization-dependent DNA cleavage domain from the FokI restriction enzyme to one of the proteins within the Cascade complex, and then target two FokI-Cascade complexes to adjacent sites in the genome to induce a single double-strand break (DSB). (Gene editing aficionados will recognize that both zinc finger nucleases and TALENs also rely on FokI dimerization to induce DSBs.)
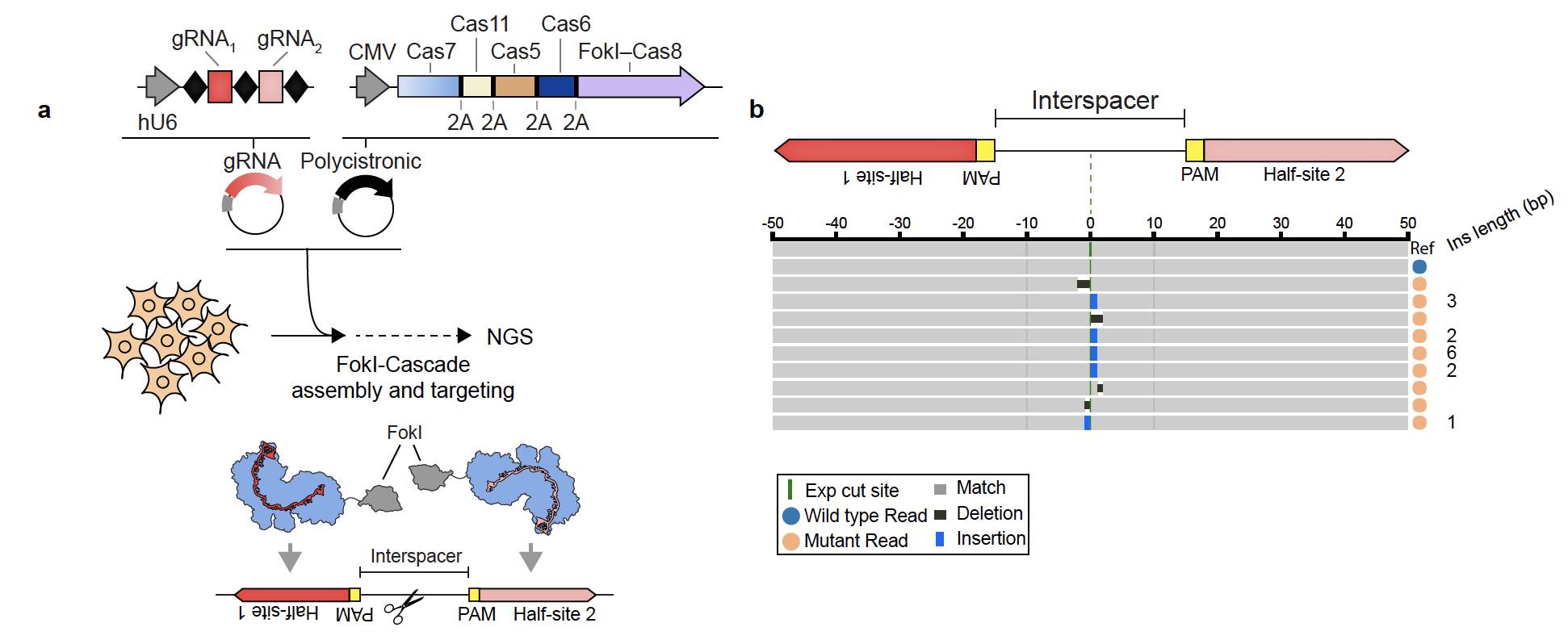
To test our design, we initially transfected HEK293 cells with purified FokI-Cascade complexes, targeting a panel of sites in the human genome. To our excitement, we observed editing efficiencies up to ~4%, which signaled to us that the strategy was working. Next, we developed a streamlined expression system comprising a 5-gene polycistronic vector and a separate PCR-generated guide RNA (gRNA) expression amplicon, allowing us to express, program, and reconstitute the full complex in cells without any need for labor-intensive ribonucleoprotein purification (see Fig. 2). Using our high-throughput cell engineering and sequencing platforms, we determined the optimal distance between the two FokI-Cascade complexes and broadly screened ortholog variants, PAM preferences, and off-target sites. In the end, we achieved editing efficiencies up to ~50% with our most active targets and with at most two off-targets across any of the guides. (For reference, a Cas9 sample run in parallel showed > 250 off-targets.) Finally, we also wondered whether our modular expression platform could be used to reconstitute the native Cas3-Cascade interference complex. Indeed, a study published during our own manuscript review process had shown that Cas3 and purified, intact Cascade complex could be co-transfected into human cells to generate long deletions (Dolan et. al., Mol. Cell, 2019 Jun 6;74(5):936-950). We were excited to observe that simply expressing Cascade protein components, Cas3, and a guide similarly resulted in targeted deletions, with lengths ranging from several bp up to ~200kb.
Now that we can harness Type I CRISPR-Cas systems in human cells, where can we go from here? Since FokI-Cascade relies on 64-nt of target sequence and has a rigid constraint for FokI positioning, there will be circumstances where it provides a significant specificity advantage over Cas9. The Cas3-Cascade system may allow new ways of screening large regulatory sequences, such as those that govern T cell exhaustion. The multiple proteins in Cascade offer many potential attachment points to tether effectors to modify the genome, such as those often used with dCas9. Indeed, another study published while our paper was in press showed how Cascade could be exploited for programmable transcriptional activation and repression (Pickar-Oliver et. al., Nat Biotechnol., 2019). Just recently, two new multi-component CRISPR transposon systems were discovered, including one which relies on Cascade, so our work may even serve as a roadmap for developing those systems in human cells (Klompe et. al., Nature, 2019 Jul;571(7764):219-225). Finally, and perhaps most importantly, multi-protein complexes are of course found in virtually every corner of biology, not just in the Class 1 CRISPR-Cas systems studied in our paper. Our work suggests that those systems are perhaps more “fair game” for biotechnology than was previously appreciated.
You can read our paper here
Follow the Topic
-
Nature Biotechnology

A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in