HK1 from HSC-derived extracellular vesicles expedites HCC progression
Published in Cancer
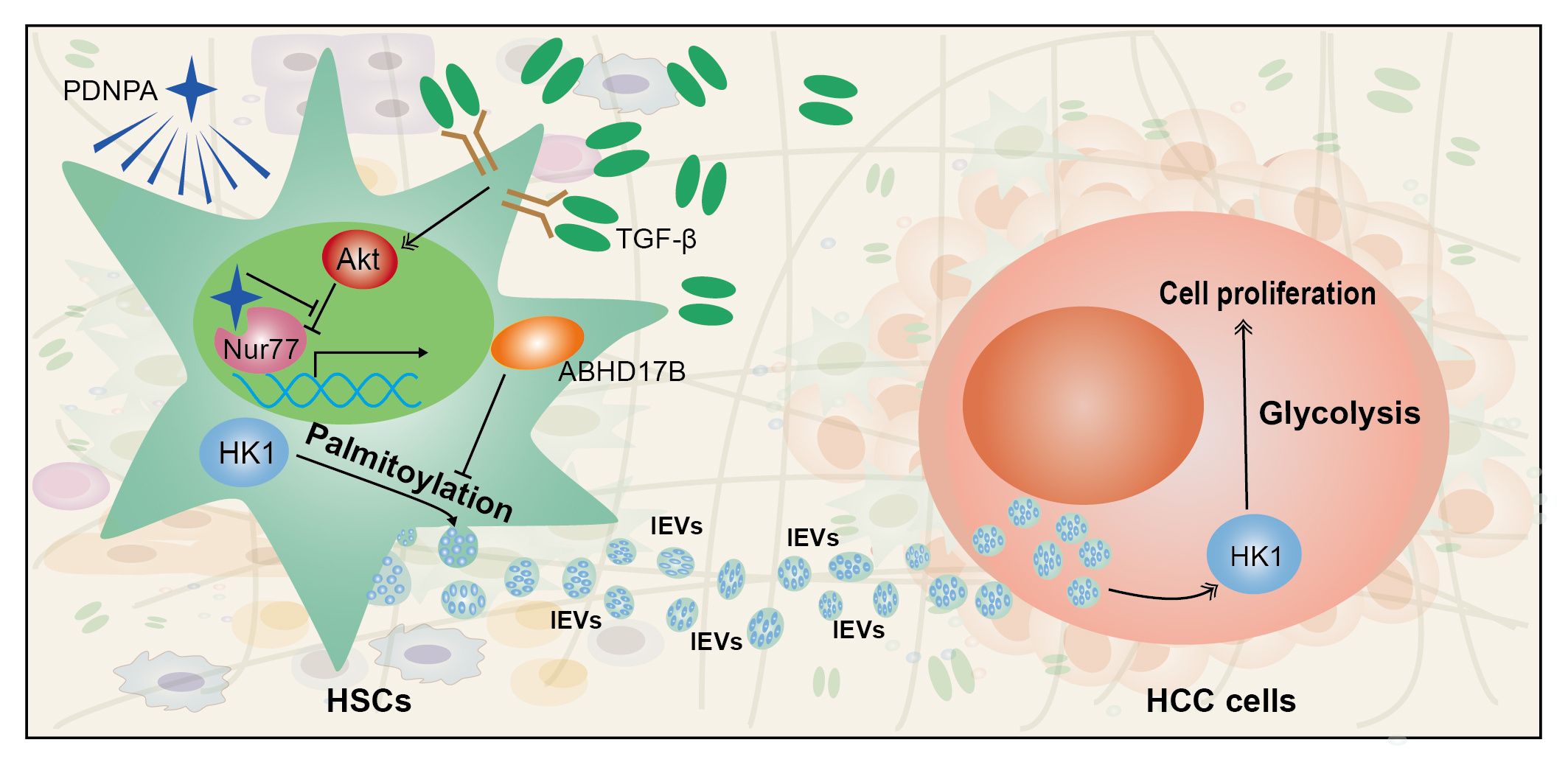

Hepatocellular carcinoma (HCC) is associated with tumor-associated stromal cells in general and hepatic stellate cells (HSCs) is the major subtype of stromal cells involved in liver fibrosis1, 2. Most HCC cases are developed from severe liver fibrosis or cirrhosis with the protumoral microenvironment in the fibrotic liver. As the most potent fibrogenic factor, TGF-β is markedly increased in the fibrotic liver to activate HSCs3. Recently, protein secretion has attracted great attention. Almost all types of cells can release extracellular vesicles (EVs) that play crucial roles in intercellular communication between tumor and microenvironment. The term “small extracellular vesicles” (sEVs) has been suggested to refer to EVs less than 200 nm in diameter, and the term “large extracellular vesicles” (lEVs) has been used to refer to EVs greater than 200 nm in diameter4.
Hepatic stellate cells play a key role in the process of liver fibrosis. We hypothecate that hepatic stellate cell-released EVs affect tumor progression. To this end, label-free quantitative proteomic analysis was initially employed to study TGF-β-induced variations in the protein profiles of the EVs in hepatic fibrosis. Through GO enrichment analysis of the differentially expressed proteins, it was found that the metabolic pathway was mostly enriched with lEVs but not sEVs. In addition, the rate-limiting enzyme in glycolysis, HK1, was highly enriched in lEVs during HSCs activation. In HSCs, HK1 located in the mitochondria under quiescent state, but was translocated to plasma membrane under the treatment of TGF-β as a prerequisite for protein sorting into lEVs. This TGF-β-induced HK1 translocation was associated with HK1 palmitoylation, a modification that assists protein targeting to the membrane5. TGF-β-induced palmitoylation of HK1 was the result of downregulating the expression of depalmitoylase ABHD17B. lEVs secretion of palmitoylated HK1 was also assisted by TSG101 in the plasma membrane. HCC cells were with a much lower level of HK1. However, their glycolysis (including glucose uptake and extracellular acidification rate (ECAR)) was evidently accelerated through the hijack of HSC-derived lEV HK1. Consequently, the uptake of lEVs led to enhanced proliferation of HCC cells. The role of lEV HK1 in promoting HCC progression was further verified in mouse models, including the orthotopic HCC model, lung metastatic experiments and DEN/CCl4-induced or HFD/STZ-induced primary HCC model. These results in vivo were consistent with the fact that lEV HK1 from the fibrotic microenvironment was hijacked by HCC cells to facilitate tumor progression.
Nuclear receptor Nur77 has been reported to be an endogenous inhibitor of fibrosis6. We found that during hepatic fibrosis, Nur77 could upregulate ABHD17B’s gene transcription and protein expression to inhibit HK1 release through depalmitoylation of HK1. However, Nur77 would be degraded in HSCs, as the result of phosphorylation by Akt under the influence of TGF-β, leading to the suppression of ABHD17B expression. It is therefore plausible that the effect of TGF-β on Nur77 could be attenuated by impeding the interaction between Akt and Nur77 to prevent HK1 secretion through lEVs. A small molecule PDNPA was identified by screening our in-house and Nur77-biased library, which restored the TGF-β-associated downregulation of Nur77 level. PDNPA bound to the ligand-binding domain (LBD) of Nur77 to generate steric hindrance to impede Akt targeting and phosphorylation. It could also be demonstrated that, in a DEN/CCL4-induced mouse model of HCC, PDNPA administration significantly suppressed the secretion of lEV HK1 from HSCs and retarded HCC development. This inhibitory effects of PDNPA on HCC development was diminished after Nur77 was specifically knocked out in HSCs.
EVs derived from cancer cells and stromal cells are considered to be one of the most important mediators of cellular crosstalk, playing vital roles in reprograming of tumor metabolism and establishment of tumor microenvironment. Our previous study demonstrated that HCC cell-derived lEV PKM2 remodeled tumor microenvironment to promote the differentiation of monocytes into tumor-associated macrophages, resulting in the malignant progression of HCC7. The current study demonstrates that lEV HK1 secretion from HSCs in fibrotic microenvironment supports HCC progression once it is uptaken by HCC cells. These works shed light on the protumoral functions of lEVs within HCC tumor microenvironment and call attention to the potential of EVs as therapeutic targets for HCC treatment.
The HK family consists of four isozymes. Among them, HK2 is mainly expressed in high levels in tumor cells with a high affinity for glucose to facilitate cell adaptation and metabolic reprogramming, which makes HK2 vital for tumorigenesis and development and an ideal cancer-specific target for HCC therapy8, 9. HK1 is expressed in most adult tissues, but its role in HCC development has long been ignored. With the in vivo and in vitro approaches, we found that HCC cells were able to hijack lEV HK1 secreted from activated HSCs to enhance cell proliferation, reflecting the resourcefulness of tumor cells. As the Michaelis–Menten constant (Km) of HK1 for glucose is much lower than that of HK210, supplement of lEV HK1 from the stromal microenvironment may not only strengthen glycolysis but also expand the scope of glucose utilization for tumors to adapt to glucose deficient situations. Since rapidly growing tumors are usually confronted with glucose starvation because of the poor vascular supply11, yet HCC is typically characterized by rapid tumor growth without hypervascularity12, it is reasonable to speculate that HSC-derived lEV HK1 may play an important role for the metabolic plasticity of HCC cells.
Nur77 was reported previously to inhibit fibrosis through the recruitment of the SP1-SIN3A-CoREST-LSD1-HDAC1 repressor complex in fibroblasts6 and our study indicated that Nur77 might suppress HK1 secretion by promoting ABHD17B-mediated HK1 depalmitoylation and inhibiting the activation of HSCs. Unfortunately, the inhibitory effect of Nur77 is abrogated by TGF-β through Akt activation in both of these instances. It is our find that the binding of compound PDNPA to the Nur77 LBD generates steric hindrance to impede the interaction between Akt and Nur77, thereby abolishing Akt-mediated Nur77 phosphorylation and degradation. So PDNPA could be a promising multifunctional lead compound that ameliorates hepatic fibrosis and suppresses HCC development.
In summary, this story demonstrates an overlooked function of HK1 in HCC upon its release from HSCs and highlights PDNPA as a candidate compound for blocking HK1 release and inhibiting HCC progression (Figure 1).
 Figure 1 The vigorous growth of tumor cells is analogous to a marching ancient army (left) which consumes large quantities of materials. Hepatic stellate cells release HK1 in material assistance to the tumor progression like porters in logistic support for the army (right). As PDNPA was employed to block the path of the porters, the support for the tumor progression will be choked off just as the movement of the army will be halted as the deficiency in material supply would impede the rapid advancement.
Figure 1 The vigorous growth of tumor cells is analogous to a marching ancient army (left) which consumes large quantities of materials. Hepatic stellate cells release HK1 in material assistance to the tumor progression like porters in logistic support for the army (right). As PDNPA was employed to block the path of the porters, the support for the tumor progression will be choked off just as the movement of the army will be halted as the deficiency in material supply would impede the rapid advancement.
Reference
- Duran, A. et al. p62/SQSTM1 by Binding to Vitamin D Receptor Inhibits Hepatic Stellate Cell Activity, Fibrosis, and Liver Cancer. Cancer cell 30, 595-609 (2016).
- Yoshimoto, S. et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499, 97-101 (2013).
- Tsuchida, T. & Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nature reviews. Gastroenterology & hepatology 14, 397-411 (2017).
- Théry, C. et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. Journal of extracellular vesicles 7, 1535750 (2018).
- Linder, M.E. & Deschenes, R.J. Palmitoylation: policing protein stability and traffic. Nature reviews. Molecular cell biology 8, 74-84 (2007).
- Palumbo-Zerr, K. et al. Orphan nuclear receptor NR4A1 regulates transforming growth factor-β signaling and fibrosis. Nature medicine 21, 150-158 (2015).
- Hou, P.P. et al. Ectosomal PKM2 Promotes HCC by Inducing Macrophage Differentiation and Remodeling the Tumor Microenvironment. Molecular cell 78, 1192-1206.e1110 (2020).
- DeWaal, D. et al. Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nature communications 9, 446 (2018).
- Pedersen, P.L., Mathupala, S., Rempel, A., Geschwind, J.F. & Ko, Y.H. Mitochondrial bound type II hexokinase: a key player in the growth and survival of many cancers and an ideal prospect for therapeutic intervention. Biochimica et biophysica acta 1555, 14-20 (2002).
- Zhang, J. et al. c-Src phosphorylation and activation of hexokinase promotes tumorigenesis and metastasis. Nature communications 8, 13732 (2017).
- Lee, S.Y. et al. Regulation of Tumor Progression by Programmed Necrosis. Oxid Med Cell Longev 2018, 3537471 (2018).
- Loong, J.H. et al. Glucose deprivation-induced aberrant FUT1-mediated fucosylation drives cancer stemness in hepatocellular carcinoma. The Journal of clinical investigation 131 (2021).
Follow the Topic
-
Nature Metabolism
This journal publishes work from across all fields of metabolism research that significantly advances our understanding of metabolic and homeostatic processes in a cellular or broader physiological context, from fundamental cell biology to basic biomedical and translational research.

Related Collections
With Collections, you can get published faster and increase your visibility.
The expanding therapeutic landscape of GLP 1 receptor agonists
Publishing Model: Hybrid
Deadline: Jan 23, 2027
Microbiome and energy metabolism
Publishing Model: Hybrid
Deadline: Dec 06, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in