How do the genomes from “clowns of the sea” differ across the North Atlantic?
Published in Ecology & Evolution


The Atlantic Puffin (Fratercula arctica) is one of, if not the, most iconic seabird of the North Atlantic. The large and colorful triangular bill combined with the puffin’s curiosity and sometimes clumsy behavior on land are unmistakable features and provide the foundation for the nicknames “sea parrot” or “clown of the sea”. The prevalence of the puffin and its integral part in our culture cannot be denied. Puffins are extremely popular as photo object (one just needs to check out the #puffin twitter handle), can be seen on a variety of stamps and bank notes, and are the official bird of the Canadian province of Newfoundland and Labrador. They have also historically been exploited for their meat and down and are still considered a delicacy in Iceland and the Faroe Islands.
Unfortunately, despite once being extremely abundant, puffins were recently designated as "vulnerable" to extinction globally and listed as “endangered” in Europe due to substantial population declines. Yet, very little is known about the cause-and-effect dynamics between population trends, ecology, the marine ecosystem, and potential threats. Similarly, despite their iconicity, the basic intra-species taxonomy (3 currently recognized subspecies) and the genetic population structure of puffins had remained unresolved. Surprisingly, the only published genetic data to date was from the early 90s and based on allozymes.

Figure 1: Atlantic puffin. Image credit – Annemarie Loof
Given the status of the puffin and the scarcity of genetic data, we decided to shed some light on the many outstanding questions, in particular from a genomic point of view. Here, I say “we” because I am extremely lucky to have worked with so many amazing colleagues (honorable mention to Bastiaan Star and Deborah M. Leigh). Together with my supervisors, Sanne Boessenkool and Kjetill S. Jakobsen, they were fundamental to the start of this project. They were always keen to help and provided extremely valuable input along the way. We decided to apply whole genome sequencing (WGS), as we realized that one needs to analyze a large number of markers to obtain insights into the genomic population structure of such a vagile seabird.
The vast majority of genetic seabird studies have used and often still use molecular techniques that are based on a few genetic markers/loci - such as allozymes, mitochondrial DNA, or microsatellites. However, this can be problematic because these markers may not have enough power to detect patterns of genetic differentiation in highly mobile species. Indeed, variation within the mitochondrial DNA, a widely used marker in genetic seabird research, is largely influenced by historical events. On the other hand, the diversity in the nuclear data better reflects contemporary factors, such as current barriers to gene flow. So, to retrieve a full picture of the genomic population structure of the Atlantic puffin and ideally gain insights into the underlying factors driving this structure, we used whole genome sequencing to be able to analyze 100,000s of loci (both mitochondrial and nuclear). This helped us to assess levels of connectivity and gene flow between distinct breeding populations and, thus, to identify relevant conservation units.
Yet, in order to apply whole genome sequencing, a reference genome is needed. Also, one needs samples that are of sufficient quality to be able to extract enough DNA and that represent birds from a large geographic range. Both have been and still sometimes can be a bottleneck to these kinds of studies. For the reference genome assembly, we had help from Martin Irestedt and the SciLifeLab, who were able to sequence and assemble a draft reference genome (which I then needed to refine further – more details can be read in the Supplement) after extracting DNA from a female puffin blood sample.
To obtain puffin samples from the various colonies around the world, we received invaluable help from the SEAPOP/SEATRACK and ARCTOX programs (I encourage to visit https://seapop.no or https://arctox.cnrs.fr/en/home/ for more information). One cannot stress enough the importance that these multinational network(s) had for the success of this project. In particular, Tycho Anker-Nilssen and Hallvard Strøm played central roles in providing us with samples (either puffin blood or feathers) and/or connecting us with other seabird researchers that could send us additional samples. Nevertheless, the work didn’t stop there. After optimizing existing laboratory protocols and making use of the in-house sequencing center at the University of Oslo (https://www.sequencing.uio.no/), we were able to analyze whole-genome sequencing data of 72 individuals from 12 colonies throughout the puffin’s breeding range.

Figure 2: Feather sample of a puffin from the Faroe Islands. Image credit – Oliver Kersten
Beyond the collaboration with members of SEAPOP, SEATRACK and ARCTOX, which was fundamental for the sampling and discussion of results, it was a workshop in Tjärnö (Sweden) in 2018 that further paved the way for the completion of this study. In this population genomics workshop organized by Pierre De Wit, I received an introduction on the analysis of population genomics datasets of non-model species, specifically with the program ANGSD. This was critical for my training for analyzing such large genomic datasets as we generated for the puffin.
In the end, it took over 2.5 years (including ca. 4 months of parental leave) to perform the labwork, analyze all of the data and place the results into their ecological context. As a result, we were able to group all of the analyzed puffin colonies into four main population clusters, which stands in contrast to the current traditional taxonomy. We also see that puffins from Spitsbergen and puffins from Norway/Iceland/Faroe, which comprise two different population clusters, interbreed on Bjørnøya and thereby form genetic hybrids. Placing the genomic differentiation into an ecological context, we propose that a complex set of drivers impacts the genetic differentiation over different spatial scales (100–1000s of km). These drivers include, but are not limited to, the geographic distance between puffin colonies, whether or not puffins from different colonies visit similar regions during the non-breeding season and therefore potentially meet, or to what extent juvenile puffins visit colonies other than the colony they were born at.
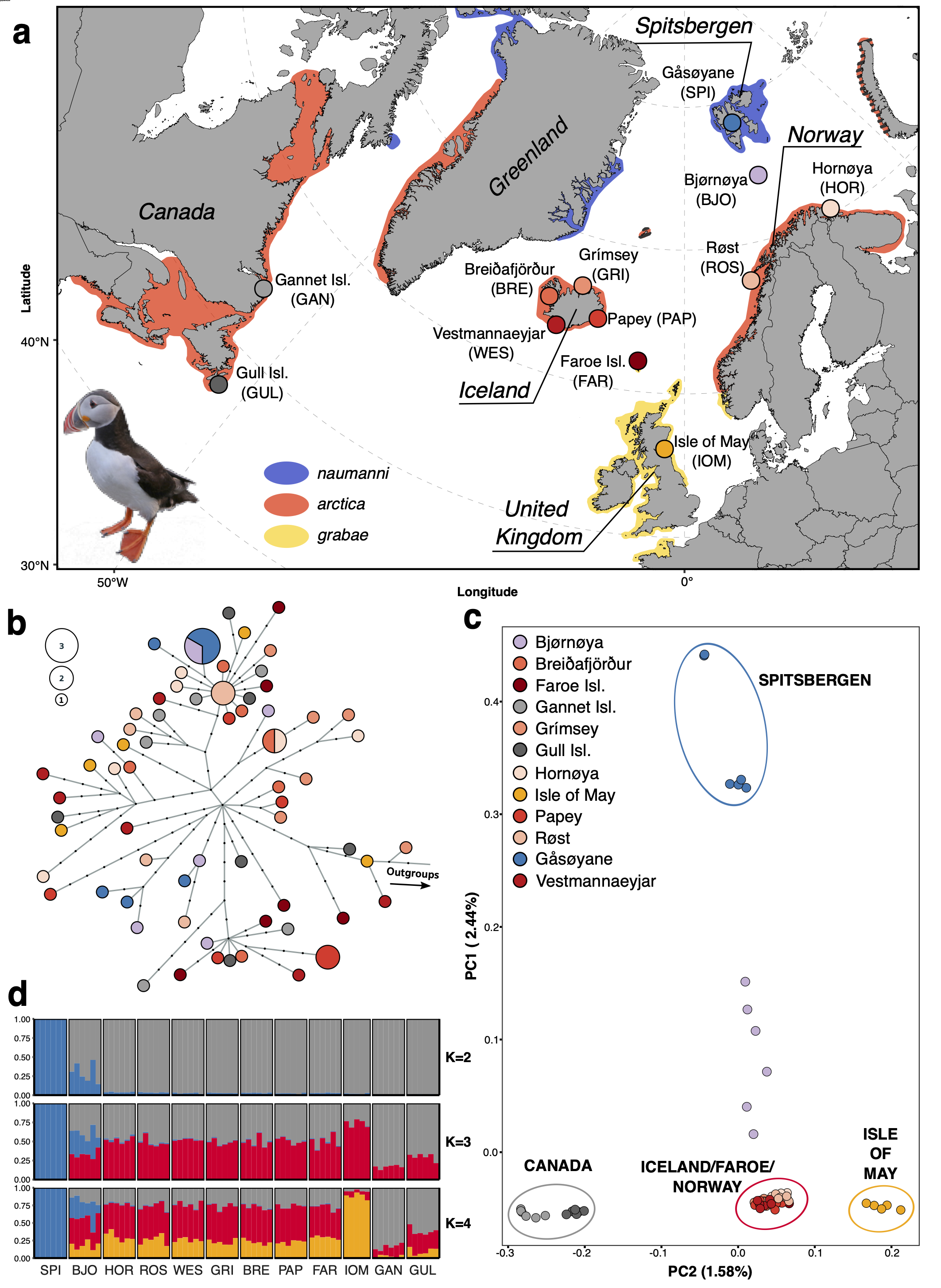
Figure 3: Figure 1 of the paper. The map in a) presents the location of the 12 sampling sites and color shading indicates the breeding range of the recognized subspecies. The color coding of the colonies is consistently used throughout the figure (a+b+c). b) A mitochondrial haplotype network based on a maximum likelihood tree. c) A principal component analysis (PCA) using genotype likelihoods to project the 71 puffins onto PC axes 1 and 2. d) Admixture plots of the best K’s using the same genotype likelihood panel as in (c).
You can have a look at our paper here!
Last but not least, stay tuned because we have only skimmed the surface of puffin population genomics and the interplay between puffin ecology and evolution. We are currently further mining this dataset. We have already sequenced representative individuals from several colonies to a higher depth of coverage and are in the process of generating a high quality “golden standard” reference assembly using PacBio, 10xG, and HiC sequencing data. Specifically, we are interested in the secondary contact zone and in the ongoing demographic and adaptive processes influencing the genomic architecture of puffins including potential structural variation. This will provide fundamental implications for our understanding of the ecology, evolution and conservation of the puffin and other seabirds with similar ecologies.
If you have further questions, feel free to send me an email or reach out on Twitter (@OKerstenScience).
Special thanks to Emma Falkeid Eriksen and Sanne Boessenkool who provided substantial contributions to this blog post.
Follow the Topic
-
Communications Biology

An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.
Related Collections
With Collections, you can get published faster and increase your visibility.
Signalling Pathways of Innate Immunity
Publishing Model: Hybrid
Deadline: Feb 28, 2026
Forces in Cell Biology
Publishing Model: Open Access
Deadline: Apr 30, 2026
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in