Human cytomegalovirus infection triggers a paracrine senescence loop in renal epithelial cells
Published in Microbiology, Cell & Molecular Biology, and General & Internal Medicine
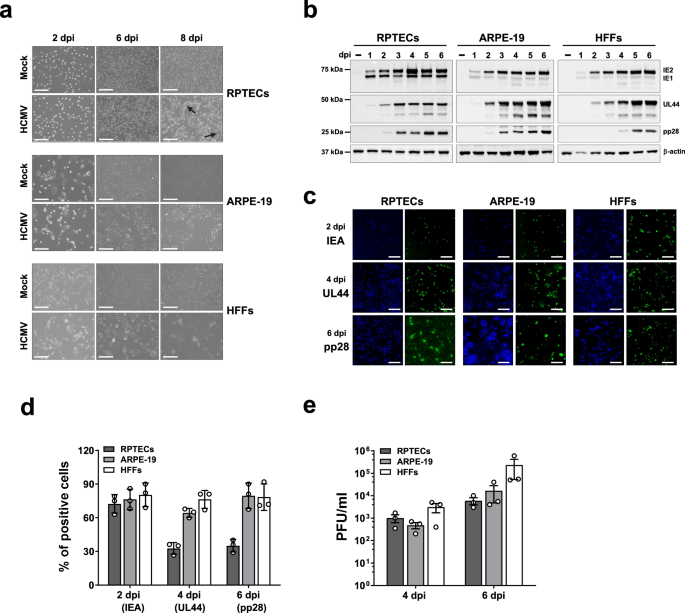
Before the COVID-19 pandemic hit, our lab was trying to understand how human cytomegalovirus (HCMV) triggers disease in specific organs. While HCMV was known to primarily infect epithelial cells in vivo, the bulk of the research on the biology of HCMV and its host interactions had been done using fibroblasts rather than epithelial cells from ocular, oral, or kidney sources—its natural hosts1. Our work, which we later shared in Communications Biology, aimed to determine whether HCMV-induced senescence contributed to organ-specific pathogenesis, using primary renal proximal tubular epithelial cells (RPTECs) as model for infection, a natural setting for HCMV disease2.
Around that time, the scientific community had begun to explore how both RNA and DNA viruses could prompt cells into a senescent state during infection3. Cellular senescence, far from being an end-stage process, serves as a critical protective mechanism, halting the division of potentially cancerous cells. When the cell senses that is in trouble due stress stimuli, including DNA damage and aging, it can enter this senescent state by arresting its cycle and secreting a cocktail of substances collectively known as senescence-associated secretory phenotype (SASP)4,5. These factors include inflammatory cytokines, growth factors, and metalloproteinase that remodel the surrounding tissue. What is really interesting is how these senescent cells can influence neighboring non-senescent cells, pushing them into a similar state of “retirement”. This process, known as paracrine or secondary senescence6, alongside the unchecked buildup and spread of senescent cells through paracrine loops, is thought to play a role in the progression of organ-specific diseases. Interestingly, this phenomenon is not just confined to naturally aging cells. Virus-infected cells also get into the act, releasing a mix of inflammatory cytokines and growth factors that are picked up by neighboring, yet uninfected cells, leading to stress amplification and damage spread7. As such, viruses, including SARS-CoV-2, are capable of inducing a unique form of cellular senescence known as virus-induced senescence (VIS) through multiple mechanisms3,8.
Due to its slow-replication rate, HCMV requires a stable cell cycle environment achieved by inducing virus-mediated cell-cycle arrest at the G1/S boundary. During this critical phase, cells are not only metabolically active but also primed for DNA synthesis, providing optimal conditions for viral replication. HCMV orchestrates this cellular environment by triggering a combination of pathways that either stimulate or inhibit the cell cycle, whose outcomes appear to be cell-type specific.
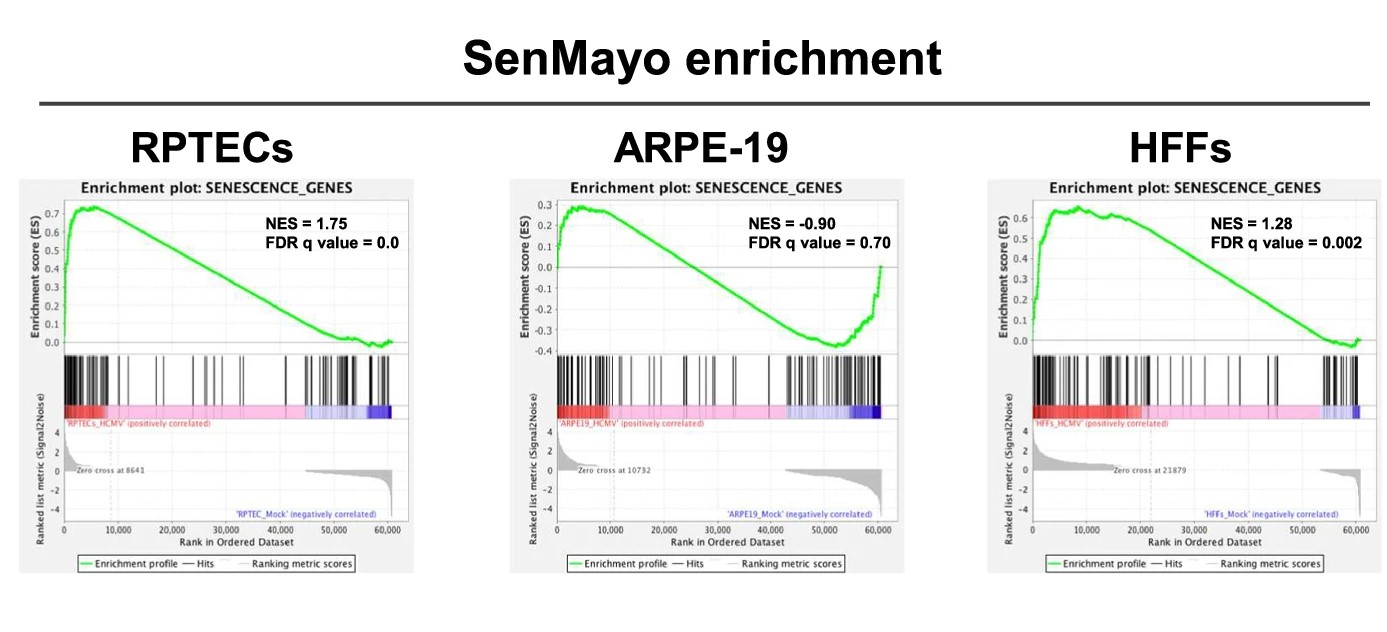
In our earlier work, we had learned that HCMV infection can make human fibroblasts enter a state of senescence, relying on a specific process involving p16INK4a 9,10. In this study, we explored how HCMV affects RPTECs using a two-pronged approach: first, by measuring the inflammatory cytokines released during infection using a Luminex® multiplex assay; and second, by identifying the main pathways modulated upon infection through transcriptomic analysis. To our surprise, we identified inflammation and organ rejection signatures that were specific to RPTECs, along with the secretion of inflammatory cytokines known to trigger senescence. Coincidentally, during this investigation, a new study introduced the SenMayo signature, a predictive marker for cellular senescence11. This provided an additional layer of validation for our findings, as we observed significant alignment of the SenMayo signature within the transcriptomic data from our HCMV-infected renal cells.

A significant breakthrough in our project occurred as we were discussing these results in a lab meeting. On that occasion, we pondered what might be the main factor involved in senescence induction and whether it could be modulated. Speculations about IL-6 came from its specific enrichment in the IL-6/JAK-STAT3 signaling pathway in HCMV-infected RPTECs, as identified by our transcriptome analysis, and from its role as a primary cytokine associated with paracrine effects. To validate our hypothesis with functional evidence, we decided to suppress IL-6 receptor signaling using tocilizumab (TCZ), a recombinant humanized anti-IL-6R monoclonal antibody commonly used to treat rheumatoid arthritis. This intervention led to reduced paracrine effects on NF-κB and H2AX activation during infection, indicating that IL-6 plays a significant role in mediating the paracrine response of these cells. Consistently, TCZ treatment of HCMV-infected RPTECs led to a dramatic decrease in the total number of bystander uninfected cells expressing senescence hallmarks, such as nuclear NF-kB and g-H2AX. Moreover, the addition of TCZ to the UVB-inactivated conditioned medium from infected RPTECs significantly impaired its ability to trigger senescence when applied to fresh target cells.
Piecing together the puzzle from our investigations, it became clear that when HCMV infects RPTECs—cells already prone to infection and linked to virus-related disease in the real world—it elicits a senescence-like program. This process halts their growth and prompts them to release inflammatory cytokines associated with aging. These cytokines, in turn, stimulate neighboring cells to enter a similar state of suspended growth.
However, one crucial question remained unanswered: is renal senescence a defensive strategy, or is it triggered by the virus to favor its own replication? Ironically, this very question was asked by one of the three Reviewers of our manuscript, prompting us to deliver a thorough and convincing response. To this end, we infected cells in presence of SASP-enriched supernatants and maintained this inflammatory component for the duration of the infection. Surprisingly, the continuous presence of this inflammatory milieu led to a decrease in the number of infected cells, implying that the senescence-associated inflammatory response may represent a mechanism through which the body tries to control or mitigate the spread of the virus, rather than the virus exploiting senescence to replicate better.

Given that this paracrine loop of disease amplification is not seen in fibroblasts and epithelial cells from non-renal tissues (e.g., retina-derived), we propose that it may be differentially relevant to specific settings, such as the kidney. This specificity constitutes the rationale for the development of rejuvenation strategies to counter VIS, which are expected to curb the spread of virus-induced damage and mitigate its associated adverse effects.
In conclusion, our research, published in Communications Biology2, demonstrated how HCMV-induced SASP in RPTECs triggers a harmful secretory phenotype, leading to the spread of paracrine senescence among the nearby uninfected cells, mainly driven by IL-6.
These results are encouraging for our multidisciplinary team, which strives to identify new molecular targets for current and future viral diseases (INF-ACT project) and seeks to address the emerging scientific and societal issues related to aging (AGING Project).
Overall, our translational research holds the potential to impact the treatment paradigm against virus-induced disease in the very near future.
References
-
Knipe, D. M. & Howley, P. Fields Virology. (Lippincott Williams & Wilkins, 2013).
-
Raviola, S. et al. Human cytomegalovirus infection triggers a paracrine senescence loop in renal epithelial cells. Commun Biol 7, 1–19 (2024).
-
Lee, S. et al. Virus-induced senescence is a driver and therapeutic target in COVID-19. Nature 599, 283–289 (2021).
-
Gorgoulis, V. et al. Cellular Senescence: Defining a Path Forward. Cell 179, 813–827 (2019).
-
Coppé, J.-P. et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6, 2853–2868 (2008).
-
Sanchez, V. & Spector, D. H. Subversion of cell cycle regulatory pathways. Curr Top Microbiol Immunol 325, 243–262 (2008).
-
Streblow, D. N., Orloff, S. L. & Nelson, J. A. Acceleration of allograft failure by cytomegalovirus. Curr Opin Immunol 19, 577–582 (2007).
-
Schmitt, C. A. et al. COVID-19 and cellular senescence. Nat Rev Immunol 23, 251–263 (2023).
-
Noris, E. et al. Cell cycle arrest by human cytomegalovirus 86-kDa IE2 protein resembles premature senescence. J. Virol. 76, 12135–12148 (2002).
-
Zannetti, C. et al. The expression of p16INK4a tumor suppressor is upregulated by human cytomegalovirus infection and required for optimal viral replication. Virology 349, 79–86 (2006).
-
Saul, D. et al. A new gene set identifies senescent cells and predicts senescence-associated pathways across tissues. Nat Commun 13, 4827 (2022).
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026
DNA repair and human disease
Publishing Model: Hybrid
Deadline: Oct 31, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in