Hybridizing chalcones and phenoxyacetamides to identify a promising lead scaffold for c-Met kinase inhibition
Published in Chemistry and Protocols & Methods
What biological problem did this study aim to address?
Aberrant activation of the hepatocyte growth factor receptor, c-Met, plays a central role in tumor growth, invasion, metastasis, and therapeutic resistance in several cancers, including breast and lung malignancies. Although multiple c-Met inhibitors have been developed, challenges related to selectivity, toxicity, and resistance remain, highlighting the need for structurally novel scaffolds with improved biological profiles.
Chalcones are α,β-unsaturated carbonyl compounds known for structural flexibility and diverse biological activity. Several naturally occurring chalcones, including butein and licochalcone B, have been reported to inhibit c-Met through hinge-region interactions, particularly involving Met1160. Separately, phenoxyacetamide derivatives have demonstrated anticancer activity across multiple tumor models, and notably, 2,4-dichloro-substituted phenoxyacetamides have shown enhanced activity in breast cancer cells.
These observations suggested that combining these two pharmacophores into a single scaffold could preserve key kinase-binding interactions while improving antiproliferative activity. This pharmacophore hybridization strategy formed the conceptual basis of the present study.
Why was the chalcone–phenoxyacetamide scaffold selected?
The α,β-unsaturated carbonyl moiety of chalcones was retained as the core pharmacophore responsible for hinge-region binding. A 2,4-dichlorophenoxyacetamide moiety was incorporated based on its documented biological relevance, ability to contribute additional hydrogen bonding and hydrophobic interactions within kinase active sites, and compatibility with the chalcone framework.
Substituents on the chalcone B-ring were systematically varied using electron-withdrawing (fluoro, chloro) and electron-donating (methoxy) groups at different positions to explore structure–activity relationships while maintaining the integrity of the hybrid scaffold.
How were the compounds evaluated biologically?
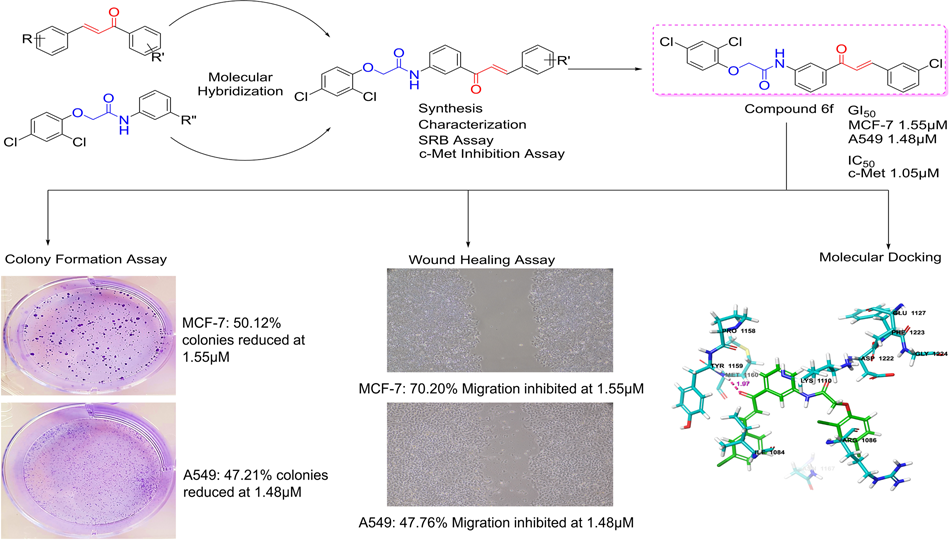
Eleven 2,4-dichlorophenoxyacetamide chalcone hybrids were synthesized using a two-step route involving amide coupling followed by Claisen–Schmidt condensation and were fully characterized by spectroscopic methods.
Antiproliferative activity was evaluated using the sulforhodamine B assay against MCF-7, A549, and HT-29 cancer cell lines. Compounds demonstrating promising cellular activity were further assessed for direct inhibition of c-Met kinase activity.
Molecular docking and molecular dynamics simulations were conducted to evaluate binding interactions and stability within the c-Met ATP-binding site. The most active compound was further evaluated using colony formation and wound healing assays to assess effects on clonogenic survival and cell migration. Docking-based kinase selectivity analysis and in silico ADME profiling were also performed.
What were the key findings?
Several compounds demonstrated low micromolar antiproliferative activity, particularly against MCF-7 cells. Among these, compound 6f showed the most consistent biological activity, including potent growth inhibition in both MCF-7 and A549 cells, strong enzymatic inhibition of c-Met, and significant suppression of colony formation and cell migration.
Structure–activity relationship analysis indicated that electron-withdrawing substituents, especially meta-fluoro and meta-chloro groups on the B-ring, were associated with improved activity, whereas methoxy substitutions were generally less favorable.
Docking and molecular dynamics studies showed that compound 6f adopted a U-shaped binding conformation and formed stable hydrogen bonds with Met1160 in the c-Met hinge region, consistent with known binding modes of class I c-Met inhibitors.
What is the significance of this work?
Hybridization of chalcone and phenoxyacetamide pharmacophores resulted in a chemically accessible scaffold with improved antiproliferative activity and favorable kinase inhibition characteristics compared with previously reported chalcone derivatives. The consistency between enzymatic inhibition, cellular activity, and computational binding behavior supports the biological relevance of this scaffold as a promising lead framework for c-Met targeting.

What are the next steps?
Future work will focus on expanding substitution patterns, refining kinase selectivity, and evaluating pharmacokinetic and in vivo antitumor properties to further optimize this scaffold.
Follow the Topic
-
Medicinal Chemistry Research
This is a journal focused on the publication of novel findings in drug design, drug discovery, and mechanisms of action of biologically active compounds.

Related Collections
With Collections, you can get published faster and increase your visibility.
Antibacterial activity
Publishing Model: Hybrid
Deadline: Ongoing
Natural products
Publishing Model: Hybrid
Deadline: Ongoing

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in