I eat (or I ate), therefore I am.
Published in Ecology & Evolution
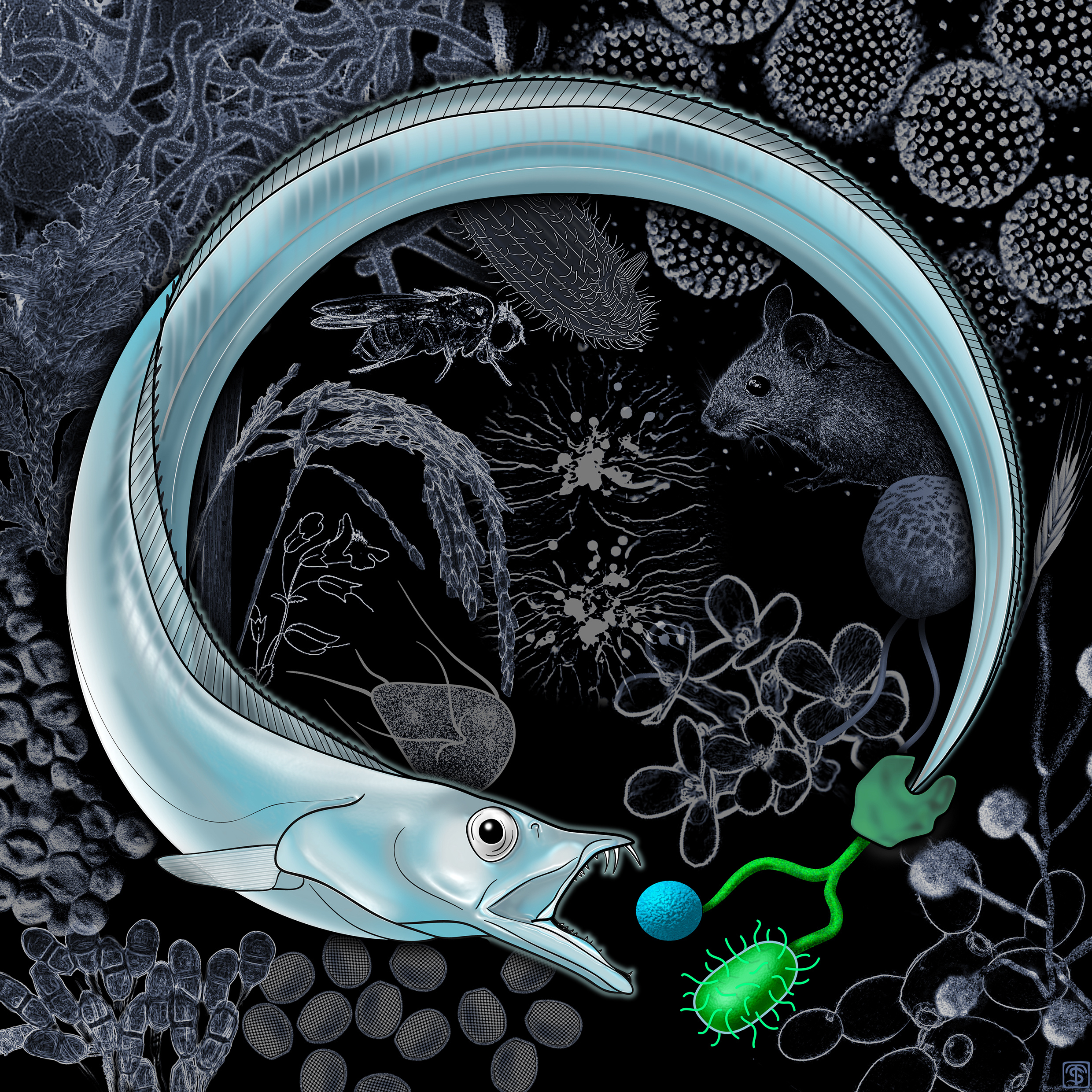
The study, in Nature Ecology and Evolution can be found here: http://go.nature.com/2BCLCYY
I am the same as a fish. A worm, a hydra, a sponge. You are too. Despite our myriad forms and niches, us animals, us metazoans, are a tiny fraction of the known diversity of eukaryotes. Just a blip. From the evolutionary perspective of a dinoflagellate or a diatom or a radiolarian, the difference between a human and a fish is very small indeed. Like gazing at a distant star, we see a point of light with no indication of the objects and planets that might be orbiting it.

To someone originally trained as a molecular biologist, like myself, this diversity is staggering. But it is also a bit like hundreds of long (like really, really long) running natural experiments. Amazingly, we are now in an age when we can access the data from those long running experiments in unprecedented detail, through genomics and computation.
I entered this project thinking about a green alga, Cymbomonas, at the prompting of my co-author Eunsoo Kim. Eunsoo and her colleague Shinichiro Maruyama showed that Cymbomonas eats bacteria.

This was a revelation because the trait of cellular eating, or phagocytosis, while suspected by some, had not been confirmed before in any plant or green alga. It is important because the ancestor of plants and green algae is a presumed phagocyte. Finding extant examples of this trait in the plants and green algae will help us better understand how plants got here in the first place. In an earlier study, we sequenced and assembled the (rather large) genome of Cymbomonas and used its genome to look for patterns of genes associated with it's "mixotrophic" lifestyle where it could both eat bacteria and use sunlight for nutrition.
In that study we found some such patterns where, using gene presence/absence, we could group Cymbomonas with both organisms that use phagocytosis, like amoebazoans and animals, and those that do not, like plants and fungi.

The patterns were there, but in that study we limited ourselves to genes present in Cymbomonas. We wondered: If we could free ourselves from the constraints of the Cymbomonas genome, would we then be able to find more general genetic patterns associated with different traits, like phagocytosis? We knew that revealing such patterns would be useful for studies on the origins of plants and green algae. We also wondered about some buzz postulating phagocytosis in new archaea that had been identified through assembled DNA sequences alone, whose cells had never been physically seen. It seemed important to define the trait of phagocytosis in a way that would allow us to make predictions on new genomes, especially those derived from mixed environmental samples.
In our first pass at finding such patterns, we grouped organisms according to their ability to use phagocytosis and then looked within those groups for shared genes that were lacking in the complementary group.

This worked to an extent, but in review the sensitivity of the approach was rightly questioned. So, we went back to the drawing board and freed ourselves from another constraint. Instead of grouping organisms by traits first, we compared all proteins in all organisms, regardless of their ability to perform phagocytosis, and used clusters of those proteins, without initial regard to the trait we were thinking about, to build sensitive protein profile models. Those models could then be used to look back for any trace of such a protein across eukaryotes, and even into bacteria and archaea.

Our expanded set of protein profile alignments spanned a reasonably large swath of eukaryote diversity, and contained patterns related to all kinds of ancient traits, including phagocytosis, photosynthesis, and small molecule biosynthesis. We pulled patterns related to those traits at the protein level and found that real predictive power came from then grouping those proteins by function and comparing functional categories across groups. But that is not the whole story. Something was missing in the analysis. We know of all kinds of phagocytotic organisms, but not all of them could be grouped using these functional patterns. It was usually parasitic organisms causing the headaches.
Parasites can tend to have relatively small, reduced/streamlined genomes, so if we wanted to learn the smallest set of proteins or functions needed for phagocytosis, we should logically look to those parasite genomes. But they just did not fit together. Different parasites seemed to have different sets of proteins related to phagocytosis and forcing them together basically broke the model. This is where a conference in Prague, Czech Republic last summer (ICOP 2017) had a real influence on the work. There is a group of organisms called the rozellids that are parasites of fungi, have reduced genomes, but are also phagocytotic. Now, I didn't really know about them early on, and I had kind of lumped the rozellids in with the fungi. One person at the conference, Guifré Torruella i Cortés, said (paraphrasing): "Absolutely not. The rozellids are definitely phagocytotic, and so are some other related organisms". I still didn't really believe him (sorry Guifré, they just fit so poorly into my model), until I saw a paper that showed, incontrovertibly, a Rozella allomycis cell with ingested mitochondria from the host fungus (Powell, Letcher, and James, 2017). That is phagocytosis.
From many passionate discussions on this during the conference, I began to form an idea. Rozella allomycis is phagocytotic, but it really didn't fit into the model with another parasite, Entamoeba histolytica. Why not make two models? In the end that worked, and from it I derived this idea that in fact, parasites do not represent the smallest set of functions needed for phagocytosis, but rather represent only the smallest set needed to fill their particular niche. They are specialists, and their genomes reflect that. With that idea, it was possible to come up with real predictive models. One representing "phagocyte generalists" that tend to be free living and have a diverse set of proteins and functions enabling them to ingest a variety of substrates under a variety of conditions, and other models representing subsets of "phagocyte specialists", who sampled from a deep ancestral pool, only retaining the proteins and functions needed to fulfill their parasitic lifestyle. It is a fascinating way to think about evolution.

Finally, because of our constraint-free approach, the protein profiles we created for this study could trace back to patterns that were likely present in the first eukaryotic cells, over 2 billion years ago. The profiles could detect related proteins in the prokaryotes, bacteria and archaea. We put together the set of proteins from our models with proteins known to be associated with the phagosome and asked which bacterial and archaeal groups have these proteins and functions? Does any broad group of prokaryotes have a set of proteins that look like a eukaryotic phagocyte? What if we stick groups together? In the end, we saw that the proteins important for phagocytosis today have varied origins from both archaea and bacteria, and no one, nor any two groups of prokaryotes could be spliced together to form a phagocytotic cell. It looks like to get phagocytosis, we needed archaea plus bacteria, plus some new proteins that are not present in any known extant group of prokaryotes.
So the paper is a story of realizing how really similar we are to one another and to all animals and how we need to understand broad diversity to understand deep evolution. Through genomics, we can now access how spectacularly diverse lineages parsed the evolutionary landscape over the course of billions of years. It is also a paper that had fits and starts, and disbelief, and what finally brought it all together was sharing ideas between friends and colleagues.
Cover image by Stephen Thurston, AMNH.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in