Inhibiting proBDNF to mature BDNF conversion leads to autism-like phenotypes in vivo
Published in General & Internal Medicine
According to epidemiological data, the prevalence of ASD worldwide has reached 1%. This disorder poses a significant threat to children's physical and mental health. If children with ASD fail to receive adequate intervention in their early years, it can lead to lifelong disabilities, affecting their physical and mental health, social interactions, learning, living, and employment. Adults with ASD often lack the ability to live, learn, and work independently, requiring long-term care and support, which places significant economic and psychological burdens on their families.
Genetic studies have revealed a close link between genetic mutations and chromosomal abnormalities and the onset of ASD. Known mutation genes associated with ASD include FMR1 in fragile X syndrome, SHANK3 in Phelan-McDermid syndrome, DHCR7 in Smith-Lemli-Opitz syndrome, UBE3A in Angelman syndrome, and CACNA1C in Timothy syndrome. A common feature of these mutated genes is that they all lead to abnormalities in the structure and function of neural synapses.
Interestingly, animal models with these genetic mutations exhibit similar core symptoms to ASD patients in experiments, such as repetitive behaviors and deficits in social interaction. More importantly, nearly all of these animal models demonstrate synaptic defects, including changes in synaptic density, abnormalities in synaptic protein synthesis, and impairments in synaptic plasticity. These findings strongly suggest a crucial conclusion: abnormalities in the structure and function of brain synapses are likely to be one of the significant factors in the pathogenesis of ASD.
Besides genetic factors, environmental factors are also considered potential triggers for ASD. For instance, prenatal infections, drug use, and other environmental exposures may increase the risk of ASD by affecting the development and function of neural synapses. Ultimately, these factors may lead to the occurrence of ASD by causing abnormalities in the structure and function of brain synapses.
Given that there is no specific drug to cure ASD, deepening its research is particularly important and urgent. This not only helps achieve early screening, diagnosis, and intervention but also optimizes patients' prognosis, improves their quality of life, reduces the burden on families and society, and lays a solid foundation for developing precision drug strategies.
Despite the significant role of genetic factors in the pathogenesis of ASD, many ASD patients do not have a clear family history of the disorder, making the research more complex. Traditional animal models of ASD, limited to specific gene mutations, are unable to fully mimic the complex diversity of ASD, thereby restricting the study of its pathogenesis and treatment strategies.
On May 18, 2024, a research paper titled "Inhibiting proBDNF to mature BDNF conversion leads to autism-like phenotypes in vivo" was published online in the journal Molecular Psychiatry by the team led by Professor Bai Lu from Tsinghua University, Professor Masami Kojima's team from Kanazawa Institute of Technology in Japan, and Professor Feng Yang's team from Beijing Tiantan Hospital affiliated with Capital Medical University. The study proposes that enzymatic defects in brain-derived neurotrophic factor (BDNF) lead to abnormalities in the structure and function of neural synapses, playing a core pathological role in the pathogenesis of ASD, marking a further step forward in understanding ASD.
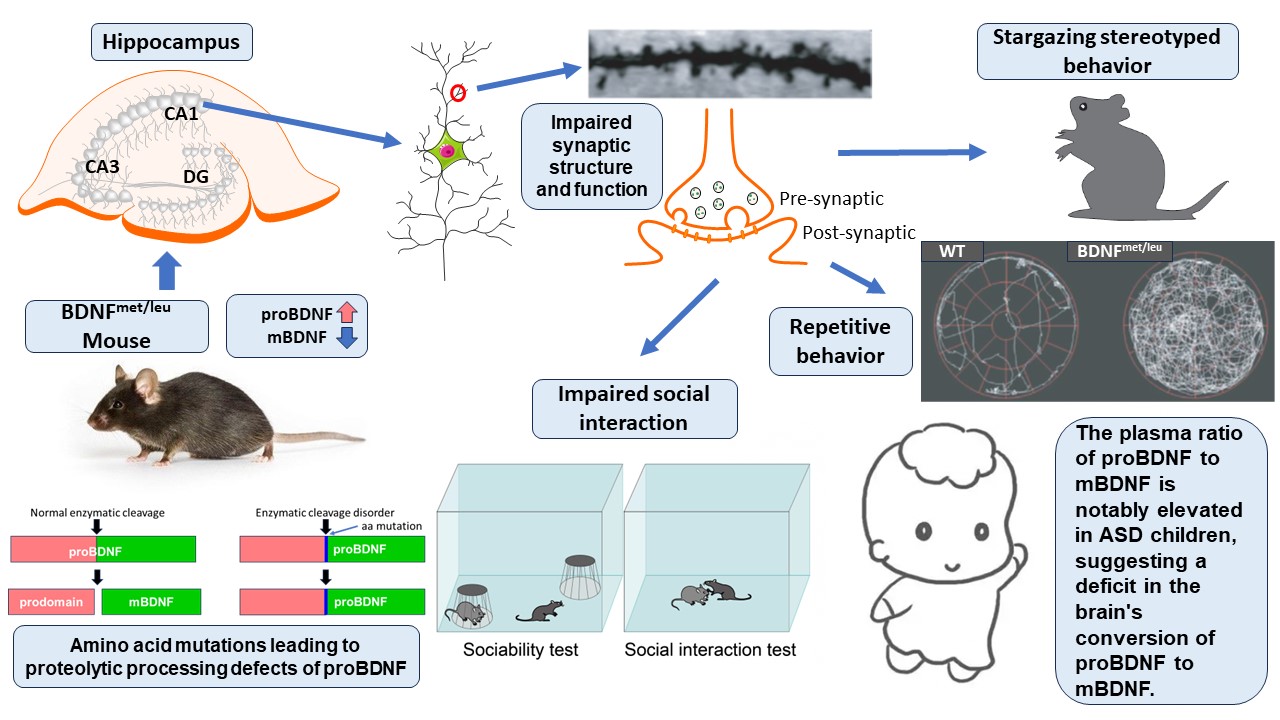
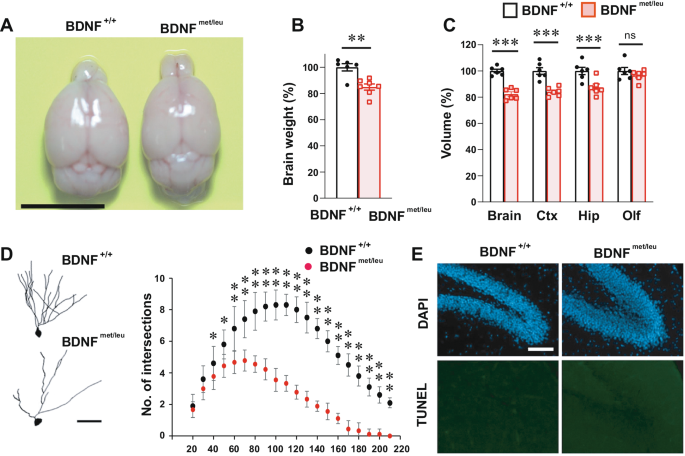
After in-depth research on the pivotal role of BDNF in neurobiology, Bai Lu's team proposed the "Yin-Yang theory" of BDNF, suggesting that proBDNF (precursor brain-derived neurotrophic factor) induces cell death and synaptic atrophy, while mBDNF (mature brain-derived neurotrophic factor) promotes cell survival and synaptic enhancement. Collaborating closely with Kojima's team, they successfully developed a unique ASD mouse model (BDNFmet/leu), which simulates the enzymatic cleavage disorder of proBDNF, resulting in a significant increase in proBDNF and a significant decrease in mBDNF production in the brain. This model exhibits synaptic impairments, deficits in social behavior, and repetitive and stereotyped behaviors similar to those observed in human ASD patients.
Compared to previous models, the BDNFmet/leu mouse model displays ASD-related phenotypes that include reduced complexity of neuronal dendrites, increased immature spines, decreased mature spines, changes in synaptic proteins, reduced synaptic transmission, and impairments in long-term potentiation (LTP) and long-term depression (LTD). These phenotypes are rarely seen in previous models, suggesting that enzymatic dysfunction in BDNF protein, rather than changes in the Bdnf gene, may be the key to the pathogenesis of ASD.
Another significant finding is that the behavioral deficits exhibited by the BDNFmet/leu mouse model are highly similar to those seen in human ASD patients, particularly the prominent repetitive behaviors such as "star gazing," a stereotyped behavior analogous to "self-stimulation" behaviors in ASD patients. This "star gazing" behavior is particularly common in ASD children, especially those with Tourette's syndrome. Although it may differ from anxiety-driven behaviors in ASD children, these behavioral phenotypes in BDNFmet/leu mice provide valuable observational indicators for pathogenesis research and drug development.
Subsequently, the team led by Feng Yang from Beijing Tiantan Hospital affiliated with Capital Medical University made significant progress in a clinical case-control study. They discovered that the levels of proBDNF and mBDNF in the plasma of children with ASD showed a significant decrease, while the ratio between proBDNF and mBDNF exhibited an upward trend. This important finding not only provides a new direction for understanding the pathogenesis of ASD, but also reveals a close connection between potential defects in the conversion process of proBDNF to mBDNF in the brain and the behavioral characteristics of ASD patients. Moreover, this discovery suggests that these plasma indicators may serve as biomarkers for ASD, providing a powerful scientific basis for monitoring disease progression and evaluating drug efficacy.
In summary, this research has thoroughly explored the phenomenon of significantly reduced mBDNF production due to proBDNF enzymatic impairment, proposing a new mechanism for the pathogenesis of ASD. This finding represents a significant advancement in the diagnosis and treatment of ASD, not only providing new directions for thinking but also laying a solid scientific foundation for monitoring disease progression and evaluating drug efficacy. It is reasonable to believe that this research will promote the further precision of ASD treatment plans and open broader paths for in-depth research in the ASD field within the medical community.
Follow the Topic
-
Molecular Psychiatry
This journal publishes work aimed at elucidating biological mechanisms underlying psychiatric disorders and their treatment, with emphasis on studies at the interface of pre-clinical and clinical research.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in