Iridium nitrenoid-enabled arene C−H functionalization
Published in Chemistry

Despite great advances in applying C−H functionalization to natural product synthesis and late-stage drug molecule functionalization, development of selective C−H functionalization protocols under mild conditions remains a fundamental challenge. In this context, metal-catalysed directed C−H functionalization allows for the highly selective activation of a specific C−H bond in the presence of other similar C−H bonds, often through the engagement of a directing group and judicious selection of a metallic catalytic system. In organic chemistry, the electrophilic aromatic substitution (SEAr) is the most common reaction type for arenes, thus dictating their functionalization reactions. Recently, a number of elegant studies describing site-selective arene functionalization that are based on SEAr were disclosed, including trifluoromethylation, fluorination, thianthrenation, borylation and fluoroalkylation. By stark contrast, only a handful of examples exist reporting direct arene C−H functionalization through nucleophilic aromatic substitution (SNAr), which can be attributed to the necessity of installing an electron-withdrawing group to facilitate the formation of a Meisenheimer intermediate, and subsequent departure of the hydride (Fig. 1a ).
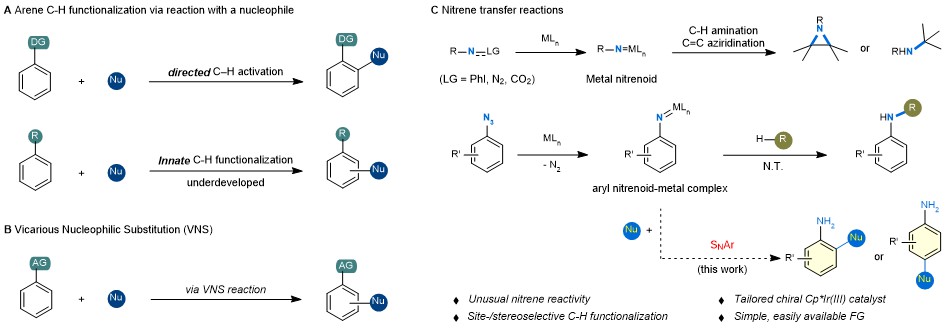
Nitrene transfer reactions represent powerful synthetic tools for the preparation of valuable amine compounds. The most common synthetic pathways enabled by nitrene transfers for the synthesis of amino organic molecules occur through either an insertion into a C−H bond or an addition to a C=C bond. In a mechanistic hypothesis, we question whether the formation of metal nitrenoid species on an arene would render the aromatic ring sufficiently electrophilic, thus allowing for an SNAr to take place. Given their ready availability, aryl azide compounds would serve as an excellent nitrene precursor. Notably, aryl azides have previously been used as nitrene precursors, but almost exclusively for the amination reaction of sp3 or sp2 C−H bonds (Fig. 1c ). We hypothesize that the formation of a putative metal nitrenoid would not only direct the incoming nucleophilic reaction partner, but also provide sufficient activation of an aryl ring to enable an otherwise not feasible SNAr.

We first examined the reaction of phenyl azide with indole as a nucleophiles in the presence of metal complexes. A survey of commonly employed transition metal complexes found that commercially available [Cp*IrCl2]2 furnished the desired product 3a in 8% yield. When cyclometalated Cp*–iridium complex Ir1 with a simple imino ligand was used, the desired product was formed in 88% yield. The generality of the reaction with regard to different aryl azides and nucleophiles was subsequently studied, and most of the substrates were converted to their corresponding products with good yields. By modifying the achiral catalyst Ir1, we eventually discovered that the chiral oxazoline-derived catalyst Ir16 can effectively promote the catalytic atroposelective synthesis of NOBIN witht readily available 2-azidonaphthalene and 2-naphthol serving as a nitrene precursor and a nucleophile, respectively.
In order to explore a possible mechanism, we conducted several control experiments, which revealed that 3,3-sigmatropic rearrangement to deliver the NOBIN products is very unlikely. 1 H NMR experiments, kinetic studies, and NLE results showed that the formation of the aryl iridium nitrenoid intermediate was the rate-determining step. DFT calculations further verified this.
For more details, please see our article: www.nature.com/articles/s41929-024-01207-3
Follow the Topic
-
Nature Catalysis

This journal brings together researchers from across all chemistry and related fields, publishing work on homogeneous catalysis, heterogeneous catalysis, and biocatalysts, incorporating both fundamental and applied studies.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in