Leveraging Cancer Modeling by Enhancing Our Ability to Extrapolate
Published in Cancer

(The author thanks co-authors Drs. Eva Perez-Guijarro and Kerrie L. Marie for discussion in concept formation, editing, and illustration)
In his remarkable book, Scale*, Geoffrey West takes us on a journey of modeling theory using as a vehicle the history of shipbuilding, which began from trial and error. He tells of the Swedish King Gustav Adolf, who in 1628 demanded to build a ship that was 30 percent longer than all others with an extra deck to carry heavier artillery. Through such radical change, West commented, “no longer would a small mistake in design lead to a small error in performance”. In her maiden voyage, the esteemed warship, Vasa, was capsized by a breeze before even leaving the harbor of Stockholm, resulting in the loss of many lives. This disaster, West explained, was caused by the fact that the contemporary ship designers did not have the scientific knowledge to scale up a ship by such a large factor. In other words, such an approach failed due to an inability to extrapolate.
This problem was later solved by William Froude around 1860. As West described: “Froude made small models of ships, three to twelve feet long, that he dragged through water contained in long tanks and measured their resistance to flow as well as their stability characteristics... He realized that the primary quantity that determined the character of their relative motion was something that later became known as the Froude number.” Further, “… because the underlying physics remains the same, objects of different sizes moving at different speeds behave the same way if their Froude numbers have the same value.”
This history is a cautionary tale that many cancer biologists would do well to heed. In fact, criticism of the clinical relevance of animal cancer models has never ceased. How to extrapolate results from animal models to human cancer is a long-standing question in the field.
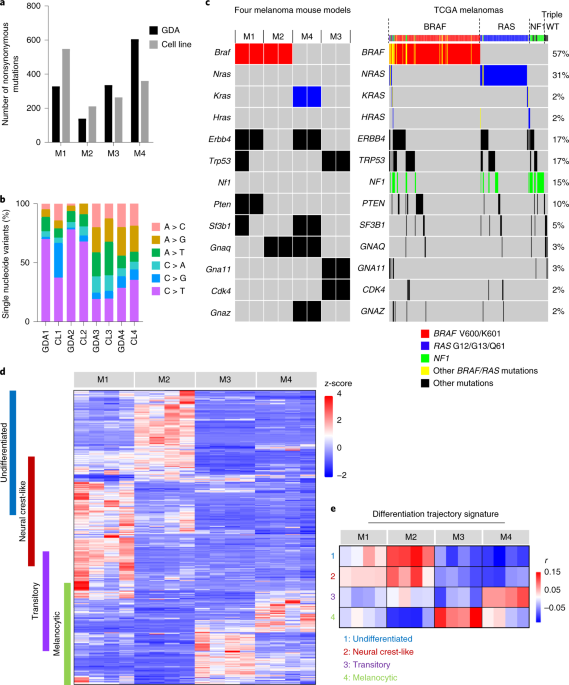
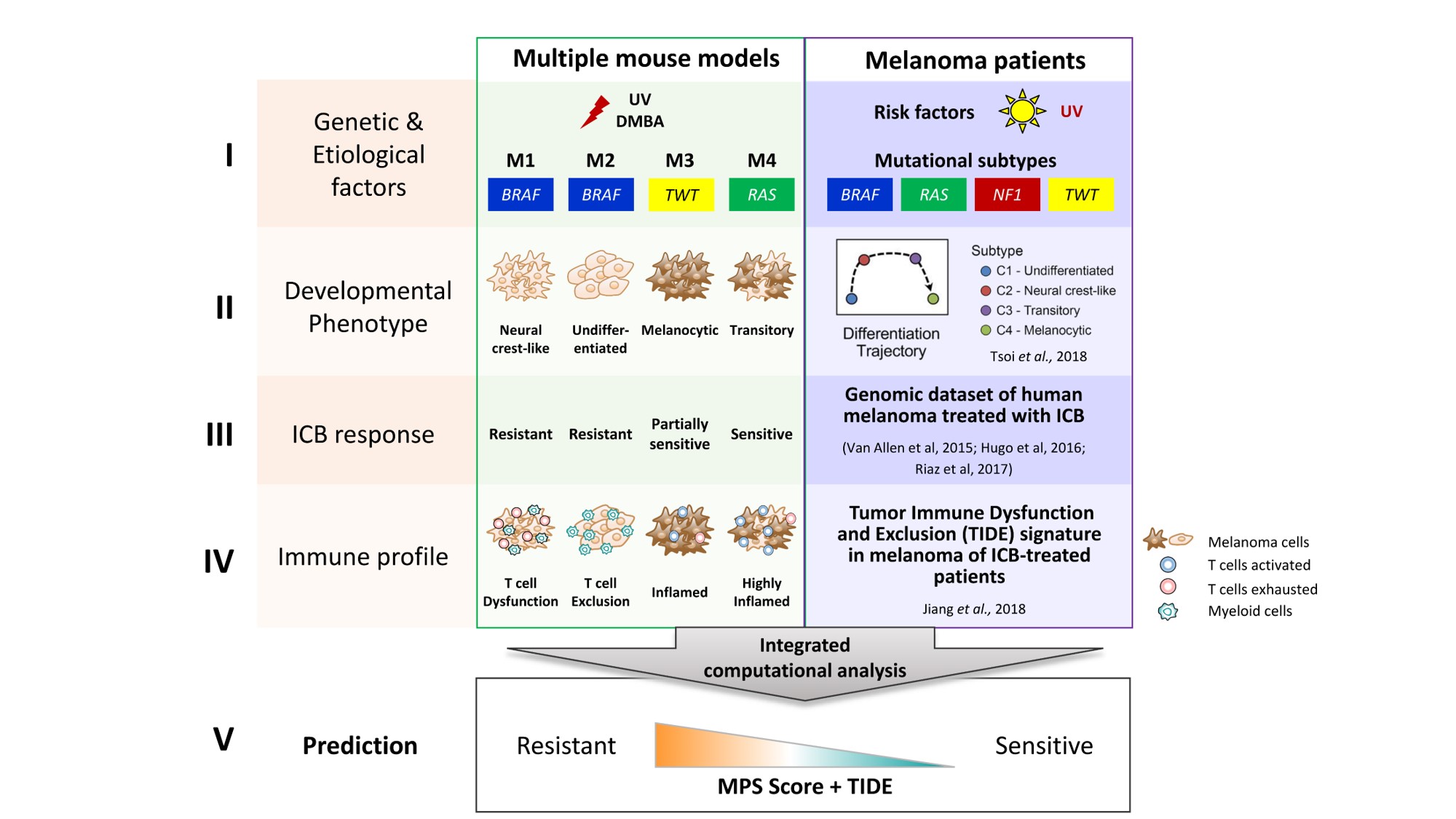
Our laboratory has been dedicated to modeling melanoma in mice. In the late 1990s, we found that neonatal UV irradiation in HGF-transgenic (tg) mice induced nodular cutaneous melanoma in 6 to 10 months. However, they lacked driver mutations of human melanoma known at that time. In the following decades we tried to improve the model by incorporating clinically relevant alleles and etiological factors at specific life stages of mice, eventually generating four major types of melanoma models, known as M1 (BrafV600E;Pten+/-;Cdkn2a+/-, UV-induced), M2 (BrafV600E;Hgf-tg;Cdkn2a+/-, UV-induced), M3 (Hgf-tg;Cdk4R24C, carcinogen-induced), and M4 (Hgf-tg, UV-induced). Now, what can we learn from these mouse models about human melanoma?
Next-generation sequencing gave us the first clue. Exome sequencing revealed that all four models exhibited distinct mutations in driver genes frequently observed in human melanoma1. From RNA sequencing, the expression differences among the four models included genes involved in development of the neural crest, the origin of melanocytes and peripheral neural tissues. Interestingly, like human melanoma2, our models can also be categorized into four developmental statuses: undifferentiated (M2), neural crest-like (M1), transitory (M4), and melanocytic (M3). In accordance, each one exhibits a distinct immune profile identified by deconvolution algorithms (e.g. Tumor Immune Dysfunction and Exclusion, TIDE3). Virtually, they are models built with different parameters.
Like Froude towed his different ship models over the test tank, we too could test our four models but with patient-relevant therapies. The results showed that M1 and M2 were resistant and M3 and M4 were sensitive to immune checkpoint inhibitors. Through a series of transcriptomic data analyses, a 45-gene signature was found to distinguish resistant versus sensitive melanomas, not only in mouse models but also in human patients4,5,6. Importantly, we found that the gene signature was associated with developmental plasticity of neural crest-derived tissues; therefore, we named it “melanocytic plasticity signature”, or MPS. Ultimately, the underlying biology that determines these therapeutic responses is associated, at least in part, with the developmental status of melanoma (https://www.nature.com/articles/s41591-020-0818-3).
Our approach revealed that results from single models cannot easily be translated to human diseases, especially when considering cross-species differences and heterogeneity in the human population. Like William Froude so elegantly demonstrated 160 years ago, effective modeling needs to test models built with different parameters, followed by a search for the underlying mechanisms that remain the same. In West’s words, the purpose of modeling is to find “dimensionless variables” allowing scaling up or extrapolating to the objects in the real world. If we consider the evolution of animals, the differences among species are still dimensional: the size of body, the metabolic rate, the time of development, etc. In this sense, the “dimensionless variable” would be the mechanisms underlying biology in body design. While Froude designed his studies based on hydrodynamics, the results of our studies demonstrated that cancer modeling should be based on evolutionarily conserved pathways, especially developmental biology. Therefore, we believe the future of cancer modeling will focus on comparative multi-model studies, whose ability to extrapolate to the clinic will be leveraged using evolutionary conserved developmental pathways. Preclinical studies of therapeutic response will be no exception.
*https://www.penguinrandomhouse.com/books/314049/scale-by-geoffrey-west/
Reference
- Akbani et al. Cell (2015), 161:1681
- Tsoi et al. Cancer Cell (2018), 33:890
- Jiang et al. Nature Medicine (2018), 24:1550
- Van Allen et al. Science (2015), 350:207
- Hugo et al. Cell (2016), 165:35
- Riaz et al. Cell (2017), 171:934
Follow the Topic
-
Nature Medicine
This journal encompasses original research ranging from new concepts in human biology and disease pathogenesis to new therapeutic modalities and drug development, to all phases of clinical work, as well as innovative technologies aimed at improving human health.

Related Collections
With Collections, you can get published faster and increase your visibility.
Clinical Research in Respiratory Medicine
Publishing Model: Hybrid
Deadline: Feb 18, 2027
Cancer Prevention and Control
Publishing Model: Hybrid
Deadline: Feb 19, 2027
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in