Long-read sequencing of the human gut microbiome
Published in Protocols & Methods
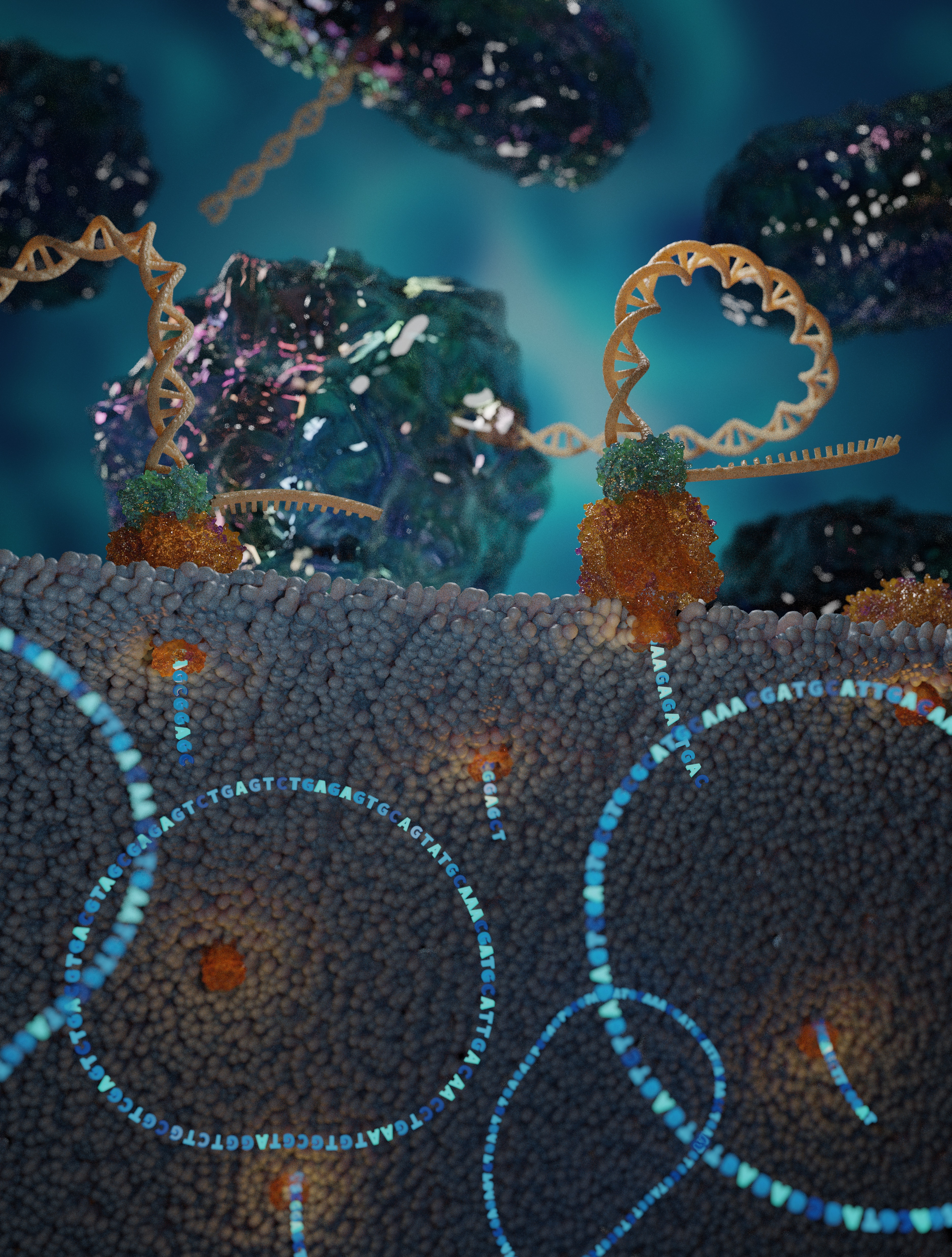
In the Bhatt Lab, we believe that strains matter. Two bacterial strains of the same species may play vastly different roles in the human gut microbiome, varying in their pathogenicity, nutrient utilization, drug response, and more1. Strain variation is generated through the acquisition of mutations and horizontal gene transfer, and can be selected for via external pressures, such as environmental variation.
Our group develops and utilizes tools to move beyond evaluating ‘Who is there?’ in a gut microbiome, and rather ask ‘What are they doing?’. Characterizing a microbiome used to rely solely on isolating and culturing bacteria from a sample. Next-generation sequencing brought us 16S rRNA and shotgun sequencing, which allow us to classify reads against a reference database. These methods give us a sense of which taxonomic groups may be present in a sample, and in what abundances. To answer the question of ‘what are microbes doing’, we need to know what the components of their genomes are. To understand this, we need to be able to build a complete picture of microbial genomes directly from a microbiome - because strains matter.
Advances in metagenomic sequencing have greatly improved our ability to directly study the genomes of bacteria without isolation and culture. Sequencing reads from a metagenome are assembled into contigs, which are then grouped through a process called ‘binning’ into unique bins that represent distinct organisms. A number of large efforts in the past few years have generated vast numbers of these metagenome-assembled genomes (MAGs)2–4, and MAGs from the human gut have been collected in the Unified Human Gastrointestinal Genome Collection5. This collection of human gut MAGs is an invaluable resource in understanding the diversity of human microbes across the globe.
However, there are inherent limitations to MAGs assembled from short-read sequencing data. Short-read MAGs are often fragmented into hundreds or even thousands of short contigs because short-read assemblers cannot correctly deconvolute the locations of repeated genomic elements, as reads are too short to span the elements and their flanking regions. These difficult-to-assemble elements include mobile genetic elements, which are commonly repeated within and across bacterial genomes and have powerful functional potential and can vary across strains of the same species.
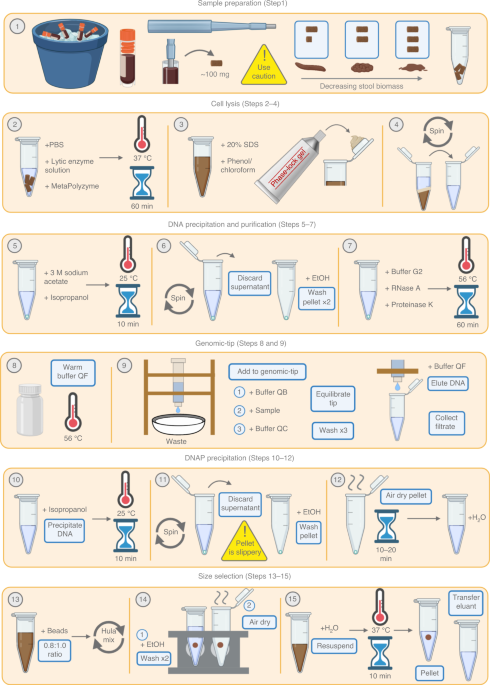
Striving to improve the contiguity of MAGs, we aimed to apply long-read sequencing technology to the human gut metagenome. This presented two challenges: first, the extraction of high-molecular weight (HMW) DNA from stool is difficult, because typical DNA extraction from stool involves vigorous bead-beating that yields highly fragmented DNA. Second, the rapid development of long-read metagenomics technology and computationally intensive steps of read processing and assembly make genome circularization from long-read metagenomic data inaccessible to non-bioinformaticians. Therefore, we developed a DNA extraction protocol that yields HMW DNA from stool, as well as a straightforward computational workflow for assembly and circularization6. Through this approach, we have generated dozens of circular or single-contig bacterial genomes that provide unique insights into the presence and potential functions of accessory genes and mobile elements in gut microbes6,7.
In this protocol piece, we aim to demystify the start-to-finish process of performing a long-read sequencing experiment on a gut microbiome sample. We include a discussion of various HMW extraction approaches, alternative computational methods, and common troubleshooting tips and tricks. We hope that these approaches will prove useful in the detailed examination of strain variation and in achieving a higher standard of bacterial genome quality.
Cover art by Ryan Brewster.
- Van Rossum, T., Ferretti, P., Maistrenko, O. M. & Bork, P. Diversity within species: interpreting strains in microbiomes. Nat. Rev. Microbiol. 18, 491–506 (2020).
- Nayfach, S., Shi, Z. J., Seshadri, R., Pollard, K. S. & Kyrpides, N. C. New insights from uncultivated genomes of the global human gut microbiome. Nature 568, 505–510 (2019).
- Almeida, A. et al. A new genomic blueprint of the human gut microbiota. Nature 568, 499–504 (2019).
- Pasolli, E. et al. Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle. Cell 176, 649–662.e20 (2019).
- Almeida, A. et al. A unified sequence catalogue of over 280,000 genomes obtained from the human gut microbiome. bioRxiv 762682 (2019) doi:10.1101/762682.
- Moss, E. L., Maghini, D. G. & Bhatt, A. S. Complete, closed bacterial genomes from microbiomes using nanopore sequencing. Nat. Biotechnol. (2020) doi:10.1038/s41587-020-0422-6.
- Tamburini, F. B., Maghini, D., Oduaran, O. H. & Brewster, R. Short-and long-read metagenomics of South African gut microbiomes reveal a transitional composition and novel taxa. bioRxiv (2020).
Follow the Topic
-
Nature Protocols
This journal publishes secondary research articles and covers new techniques and technologies, as well as established methods, used in all fields of the biological, chemical and clinical sciences.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in