Closing bacterial genomes from the human gut microbiome using long-read sequencing
Published in Bioengineering & Biotechnology

In our lab, we focus on the impact of the gut microbiome on human health and disease. To evaluate this relationship, it’s important to understand the particular functions that different bacteria have. As bacteria are able to exchange, duplicate, and rearrange their genes in ways that directly affect their phenotypes, complete bacterial genomes assembled directly from human samples are essential to understand the strain variation and potential functions of the bacteria we host. Advances in the microbiome space have allowed for the de novo assembly of microbial genomes directly from metagenomes via short-read sequencing, assembly of reads into contigs, and binning of contigs into putative genome drafts. This is advantageous because it allows us to discover microbes without culturing them, directly from human samples and without reference databases. In the past year, there have been a number of tour de force efforts to broadly characterize the human gut microbiota through the creation of such metagenome-assembled genomes (MAGs)[1–4]. These works have produced hundreds of thousands of microbial genomes that vastly increase our understanding of the human gut. However, challenges in the assembly of short reads has limited our ability to correctly assemble repeated genomic elements and place them into genomic context. Thus, existing MAGs are often fragmented and do not include mobile genetic elements, 16S rRNA sequences, and other elements that are repeated or have high identity within and across bacterial genomes.
Our lab has long been interested in creating more complete and contiguous “de novo reference genomes” directly from microbial mixtures. Our first somewhat successful attempts at this leveraged “linked read” or “read cloud” library preparation methods – and did, indeed, result in much more contiguous genomes [5]. In some cases, we improved genome contiguity, which is measured by a metric called “N50” by 10-fold. One major advantage of this method was that it only required 1 ng of DNA input. Unfortunately, there were cases in which this method did not result in such improvements. For example, many short-read and linked-read attempts to assemble Prevotella copri, a highly prevalent microbe that has been linked to diet-dependent health effects [6], have failed due to its repetitive genome. We were keen to find alternative technologies that could aid in this effort.
Long read sequencing offers a new path towards improved microbial genome assembly, as demonstrated previously [7,8], because long reads can span the lengths of these repeated elements. However, it requires much more DNA (100 ng – 1 ug) and extraction of high molecular weight DNA from stool samples is challenging due to the vigorous mechanical lysis used in standard extraction approaches. To overcome this, we adapted enzymatic lysis protocols to create a refined approach for the extraction of high molecular weight, pure DNA suitable for nanopore sequencing. Then, we developed a complementary computational workflow, Lathe, to perform long-read assembly and genome circularization from nanopore data.
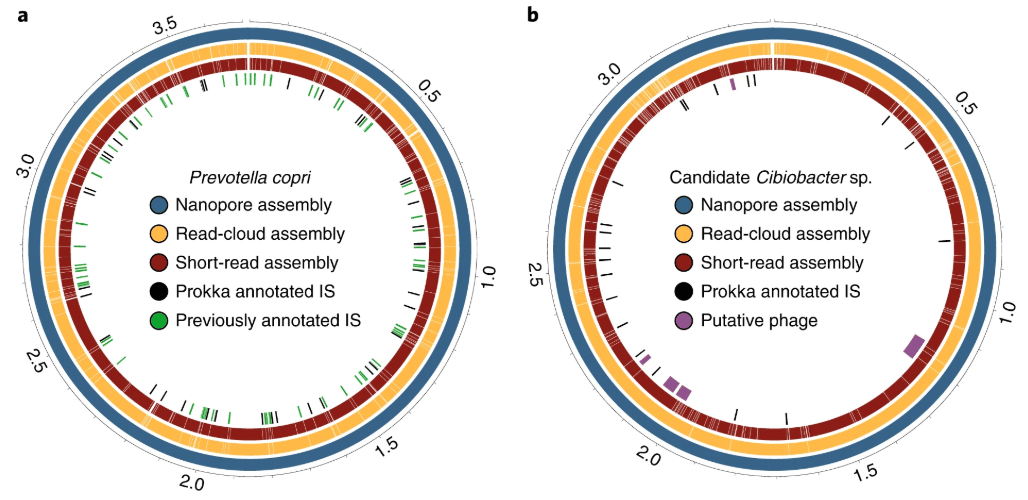
After validating the utility of our approach on mock bacterial communities, we went on to apply our method to human stool samples. We generated twenty circularized bacterial genomes and several additional high quality genomes that span a wide range of phylogeny and genome sizes. We successfully assembled a circular genome for P. copri, and subsequent annotation of its genome identified at least 69 transposases, genes that are often highly repetitive within and across bacterial genomes – a clue as to why it had eluded previous assembly attempts. We were excited to discover that an additional circular genome appears to be a candidate Cibiobacter sp., representing the first closed genome of this newly described prevalent clade [1].
The ability to generate circular bacterial genomes directly from the gut microbiome will enable accurate mapping of horizontally transferred elements, which in turn offers important insight into variation in bacterial phenotypes. We hope that our proposed approach, in combination with advancements in metagenomic sequencing and assembly algorithms, will lead to a new standard of bacterial genome quality.
Cover photo credit: Ryan Brewster
1. Pasolli, E. et al. Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle. Cell 176, 649–662.e20 (2019).
2. Nayfach, S., Shi, Z. J., Seshadri, R., Pollard, K. S. & Kyrpides, N. C. New insights from uncultivated genomes of the global human gut microbiome. Nature 568, 505–510 (2019).
3. Almeida, A. et al. A new genomic blueprint of the human gut microbiota. Nature 568, 499–504 (2019).
4. Almeida, A. et al. A unified sequence catalogue of over 280,000 genomes obtained from the human gut microbiome. bioRxiv 762682 (2019) doi:10.1101/762682.
5. Bishara, A. et al. High-quality genome sequences of uncultured microbes by assembly of read clouds. Nat. Biotechnol. 36, 1067–1075 (2018).
6. De Filippis, F. et al. Distinct Genetic and Functional Traits of Human Intestinal Prevotella copri Strains Are Associated with Different Habitual Diets. Cell Host Microbe 25, 444–453.e3 (2019).
7. Stewart, R. D. et al. Compendium of 4,941 rumen metagenome-assembled genomes for rumen microbiome biology and enzyme discovery. Nat. Biotechnol. 37, 953–961 (2019).
8. Bertrand, D. et al. Hybrid metagenomic assembly enables high-resolution analysis of resistance determinants and mobile elements in human microbiomes. Nat. Biotechnol. 37, 937–944 (2019).
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in