Loss of LECT2 promotes ovarian cancer progression by inducing cancer invasiveness and facilitating an immunosuppressive environment
Published in Cancer
Epithelial ovarian cancer (EOC) remains one of medicine’s greatest challenges to screen and target, with approximately 70% of its patients experiencing malignant ascites and intraperitoneal metastasis at staging laparotomy. Despite initiatives utilizing immune checkpoint blockade antibodies, the suboptimal response rates in EOC indicate the elusiveness of this therapeutic strategy.
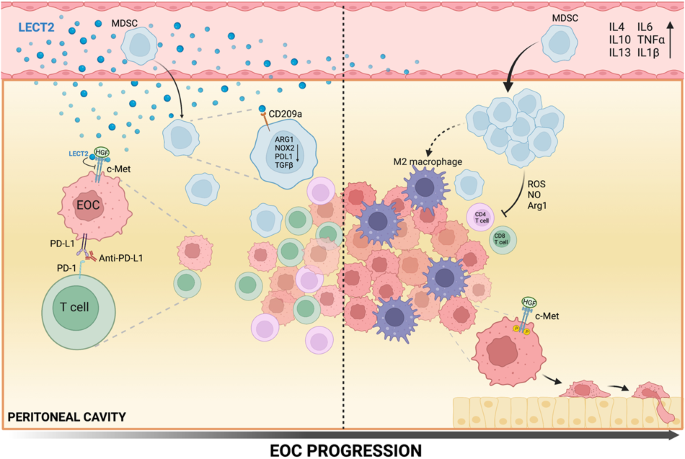
Human leukocyte cell-derived chemotaxin 2 (LECT2) is a protein secreted in the blood that is primarily found in the cytoplasm of liver cells before secretion. This protein has been found on a specific locus of chromosome 5 that holds several genes encoding for immunomodulatory cytokines. We observed and demonstrated LECT2’s various distinct functions and tumor-suppressive role in the progression of EOC, highlighting its potential for developing a novel and successful therapeutic strategy for EOC.
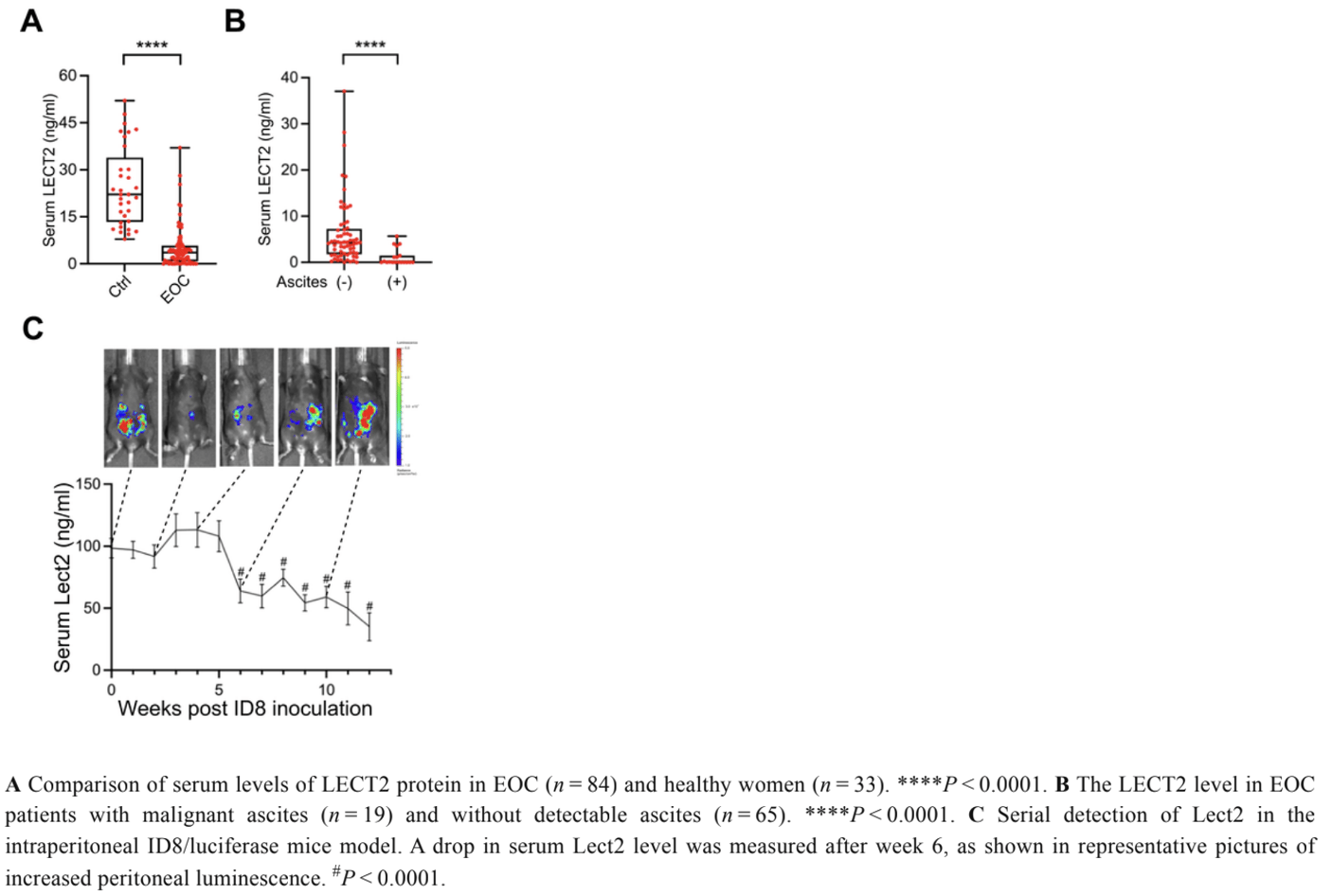
The amount of circulating LECT2 in the serum of 83 EOC patients was compared with that of 33 healthy volunteers. Our findings reveal a significant difference in circulating LECT2 levels between the two groups: those with EOC had much lower levels of serum LECT2 than the control, and those with malignant ascites had even lower levels than the EOC patients without ascites. In the examination of LECT2 serum levels using an ID8 syngeneic mouse EOC model, the stability of LECT2 concentration persisted during the first five weeks post-injection of ID8/Luc cells. However, a deviation occurred in the 6th week, marked by a significant decrease in LECT2 serum levels. These results demonstrate reduced expression of LECT2 in EOC patients and an increase in down-regulation of LECT2 as the disease progressed in the mouse model.
Further, the tumor burden and amount of ascites, known to be associated with the advanced state of EOC, in LECT2-deficient mice (Lect2-/-) were found to be higher than the wild-type (Lect2+/+) and heterogeneous (Lect2+/-) mice. The tumors in Lect2-/- mice were also larger and had metastasized to surrounding tissues. In mice of the Lect2+/+ genotype injected with ID8/Luc cells that were manipulated to overexpress LECT2, there was a diminished tumor burden and fewer ascites than the wild-type control group. Together, the findings indicate the importance of LECT2 in suppressing the progression of EOC.
Another facet to LECT2’s functionality is its ability to inhibit c-Met signaling and reduce expression of c-Met. Overexpression of c-Met is seen in various cancers such as those affecting the ovary, breast, lung, colon, and pancreas. We observed and determined that recombinant LECT2 proteins effectively suppressed the adhesion potential of EOC, SKOV-3, and ID8 cells. It was also noted with Western blot and immunohistochemical staining that phosphorylated c-Met levels and downstream signaling molecules in the c-Met pathway were higher in tumors removed from Lect2-/- mice than the Lect2+/+ and Lect2+/- mice. We targeted this signaling pathway with Cabozantinib, which inhibits c-Met. Treatment with Cabozantinib showed that ID8 tumor burden and seeding were inhibited or eliminated in Lect2-/- mice. Thus, LECT2 is essential in suppressing c-Met signaling and EOC’s aggressive adhesive and metastatic properties.
As LECT2 levels decline during the progression of the disease, a simultaneous increase in immunosuppressive and pro-inflammatory cytokines is observed, suggesting that LECT2 has anti-inflammatory properties. Loss of LECT2 revealed many tumor-promoting factors. For instance, lowered levels of LECT2 enriched ascitic CD206+ TAMs (tumor-associated macrophages) in the EOC model. Our data supports that CD206+TAMs contribute to the heightened formation of ascites in Lect2-/- mice and accelerate ID8 tumor growth. They are thus associated with EOC tumor angiogenesis. In addition, the loss of LECT2 was determined as a contributing factor to the polarization of M2-subtype TAM, potentially playing a role in metastasis. TAMs have been shown to aggravate immunosuppression through the expression of certain immune checkpoints. Notably, our investigation has revealed that LECT2 induces alterations in gene expression of MDSC, suggesting that LECT2 can inhibit MDSC-mediated T-cell suppression by interacting with the receptor CD209a. MDSC, or myeloid-derived suppressor cell, play a crucial role in hindering T-cells function and drive immunosuppression in EOC. Our data substantiates this by demonstrating that in Lect2-/- mice, MDSC enriches the ascites microenvironment, suggesting that MDSC acts as a precursor to TAMs. Thus, loss of LECT2 leads to a chain reaction of events promoting tumor progression: MDSC accumulates, potentially triggering TAMs and pro-inflammatory cytokines that diminish T cells abilities to fight the cancer. Finally, the loss of LECT2 reduces the effective blockade of the PD-1 pathway. Normally, blocking this pathway allows for a more efficient immune response in EOC. However, our data revealed that in Lect2-/- mice, the blockade of PD-1 was significantly reduced, allowing for tumor progression.
In summary, we have determined that LECT2 is crucial in the suppression of tumor progression in EOC. The loss of LECT2 results in serious implications for cancer progression and negatively impacts the success of immunotherapy. While further research is needed, these findings support LECT2 as having a strong potential in future immunotherapeutic strategies.
Follow the Topic
-
Oncogene
This journal aims to make substantial advances in our knowledge of processes that contribute to cancer by publishing outstanding research.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in