Machine Learning Unlocking Elusive High-Pressure Phase of Sodium
Published in Physics
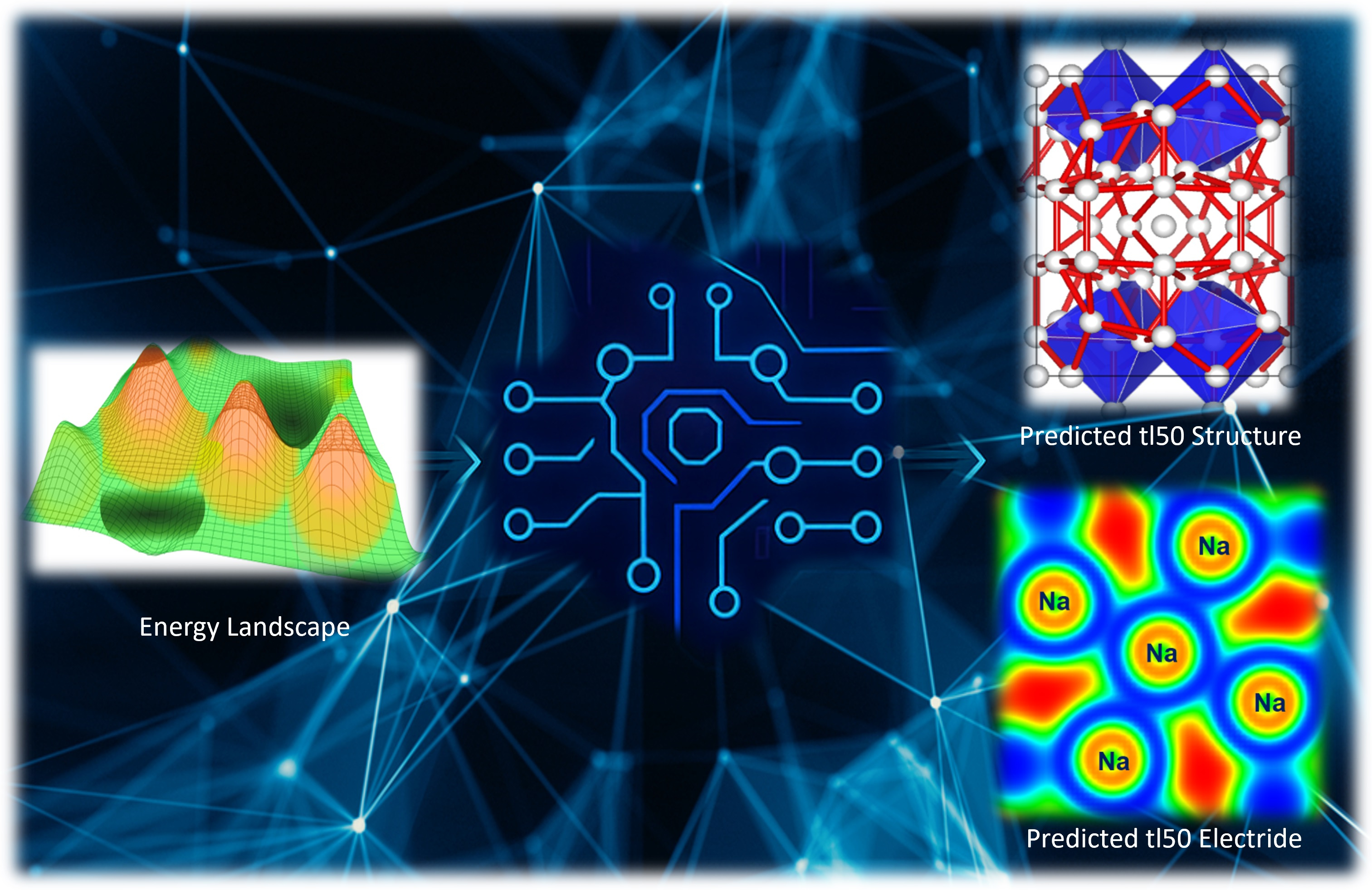
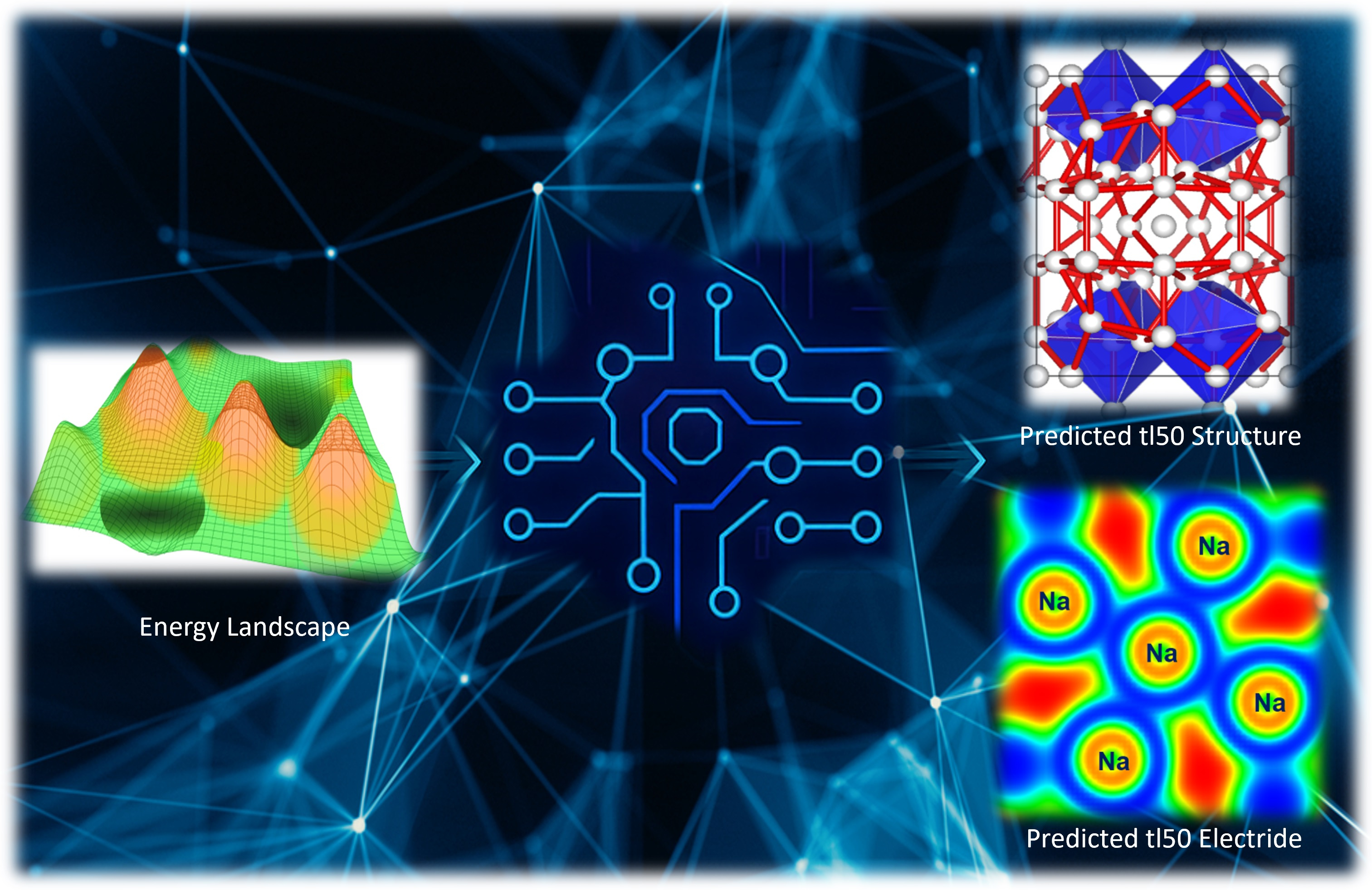
The long-held belief that metals would prefer densely packed structures with nearly free electrons (NFE) has been fundamentally challenged by high-pressure experiments. At extreme pressures, compression can cause core-valence or even core-core overlaps, which can result in interesting and unexpected changes in the electronic characteristics of metals. This phenomenon is especially noticeable in alkali metals like Li and Na, where such overlaps are expected to result in an insulating state with a narrow band gap, a prediction that has been experimentally confirmed. Despite the intuitive expectation that compression would increase interatomic interactions and broaden the electronic bands, making alkali metals even more free-electron-like, research has shown that these elements adopt remarkably complex structures under high pressure. At room temperature, elemental sodium in particular shows a number of pressure-induced phase changes.
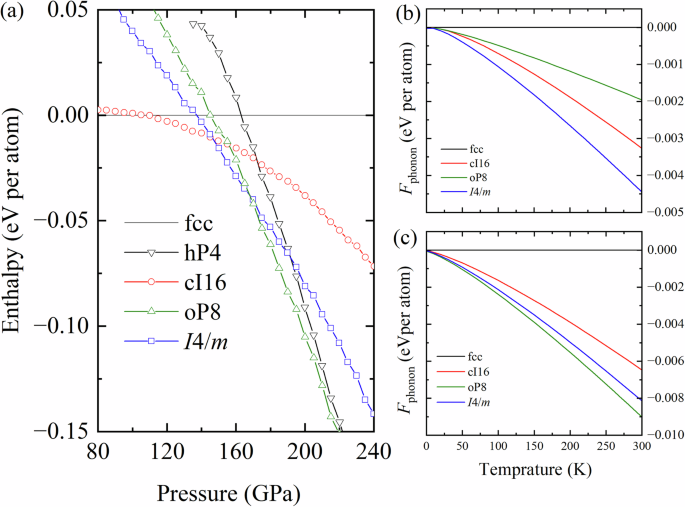
In 2008, Gregoryanz et al. (Science 320, 5879 (2008)) made a critical discovery: the minimum of the melting curve coincides with the presence of seven distinct crystalline phases identified through single-crystal diffraction experiments. To date, three of these phases (cI16, oP8, and tI19) have been fully characterized, one of which is also observed in Li under high pressure. However, the remaining four phases (tl50, aP90, oC120, and mP512) remain unresolved due to their structural complexity with large numbers of atoms in the unit cells (as indicated by their Pearson notations).
The roadblocks of traditional approaches
The standard approach for crystal structure prediction and characterization is via density functional theory (DFT)-based crystal structure searches. Herein, random crystal structures are generated with constraints, and DFT is employed for local optimization. This is generally followed by a fitness test in which high-enthalpy structures are discarded. The process continues if the enthalpy values remain unchanged.
However, traditional DFT-based crystal structure searches are hampered first and foremost by sheer computational expense and the combinatorial explosion of possible configurations. Even moderate unit cells with a few dozen atoms require many thousands of individual relaxations, and since DFT scales as 0(N3) with system size, exploring larger or more compositionally complex systems becomes prohibitively slow. On top of this, global optimization algorithms—whether genetic, particle-swarm, or minima-hopping—must sample an astronomically large potential-energy surface. Additionally, searches can get trapped in familiar basins, limiting discovery to slight variants of known motifs.
Overcoming Challenges
The bottleneck presented above is one of the reasons why four of the seven distinct crystalline phases identified through single-crystal diffraction experiments by Gregoryanz et al. in 2008 remained elusive. To overcome these barriers, our recent publication in Communications Chemistry presents an investigation into the long-elusive structures of Na observed by Gregoryanz et al. using state-of-the-art computational techniques. A comprehensive search for Na structures was conducted at 120 GPa, near where the complex phases were experimentally observed by employing machine learning assisted crystal structure approach. The search is carried out across a polymorphic space of 250,000 randomly generated structures, incorporating unit cells with up to 120 atoms in all possible space groups using in which the randomly generated structures were optimized using the constructed machine learning interatomic potential (MLIP). Details of the structure search are presented in our paper in Communications Chemistry. We confidently report the tI50 structure as a body-centered tetragonal structure with the I4/m space group. Likewise, topological analysis of the electron density confirms this structure to be an electride.
Conclusion
Elemental sodium is a metallic material. The delocalized nature of metallic bonding poses a significant challenge for structure predictions based on machine learning potentials that are implemented based on local bond topology. Despite this, we were able to construct a sodium machine learning interatomic potential (MLIP) to uncover the crystal structure and properties of the elusive tI50 phase, first observed experimentally fifteen years ago. The difficulty -- or conversely, the success––of predicting metallic structures using our approach serves as a compelling validation of MLIP capabilities. Additionally, resolving this unique structure provides new insights into sodium’s diverse structural behavior under high pressure.
Learn more
To dive deeper into the theory, methodology, and results, check out our full paper in Communications Chemistry: https://doi.org/10.1038/s42004-025-01566-3
Follow the Topic
-
Communications Chemistry
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the chemical sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Chemical modification of proteins
Publishing Model: Open Access
Deadline: Sep 30, 2026
Sustainable waste management through polymer upcycling
Publishing Model: Open Access
Deadline: Aug 31, 2026





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in