Magnons from time-dependent density-functional perturbation theory and nonempirical Hubbard functionals
Understanding the magnetic behavior of materials has long been a fundamental challenge in condensed matter physics, not only for its intrinsic scientific interest but also for its pivotal role in shaping future information and energy technologies. At the heart of this effort lies the quest to accurately model spin excitations—collective fluctuations of magnetic moments that give rise to quasiparticles called magnons. These quasiparticles are central to fields as diverse as spintronics, quantum computing, and magnonics.
But in practice, modeling magnons in real, complex materials—particularly those containing strongly localized electrons such as in transition-metal or rare-earth compounds—has been far from straightforward. Traditional computational approaches often rely on empirical parameters or simplified spin models that lack full predictive power and generality.
In our recent publication in npj Computational Materials, we present a novel first-principles methodology that opens the door to highly accurate, fully nonempirical predictions of spin-wave spectra. The approach combines time-dependent density-functional perturbation theory (TDDFPT) in the Liouville-Lanczos (LL) framework with Hubbard-corrected density-functional theory (DFT+U)—where the Hubbard U parameters are computed ab initio and treated self-consistently. This fusion offers a new, robust route for capturing magnetic excitations in materials where conventional approximations often falter.
The roadblocks of traditional approaches
The standard method for modeling magnons has long involved the use of Heisenberg spin Hamiltonians. In this picture, the collective spin dynamics of a material are encoded in interatomic exchange parameters (typically denoted J), which govern how spins interact across the crystal lattice. These J values are either empirically fitted to experimental spectra or computed from first-principles through total energy differences or perturbative techniques.
However, this framework has limitations. The J parameters are highly sensitive to the value of the Hubbard U, which is often chosen semi-empirically or without careful cross-validation. Additionally, the Heisenberg approach has challenges when it comes to noncollinear magnetic configurations, low-symmetry systems, or materials where itinerant and localized electrons coexist. Crucially, it cannot account for more subtle effects such as Landau damping or chiral spin textures, which are increasingly relevant in modern material platforms like altermagnets, skyrmions, and low-dimensional magnets.
A new path: TDDFPT meets nonempirical DFT+U
To overcome these barriers, we built a new computational framework that directly evaluates the dynamical spin susceptibility—the central quantity governing the response of a material's spin density to an external magnetic perturbation. Our approach is rooted in time-dependent density-functional theory (TDDFT), specifically its density-functional perturbation theory flavor, which solves the linear response of the system in a perturbative and efficient manner.
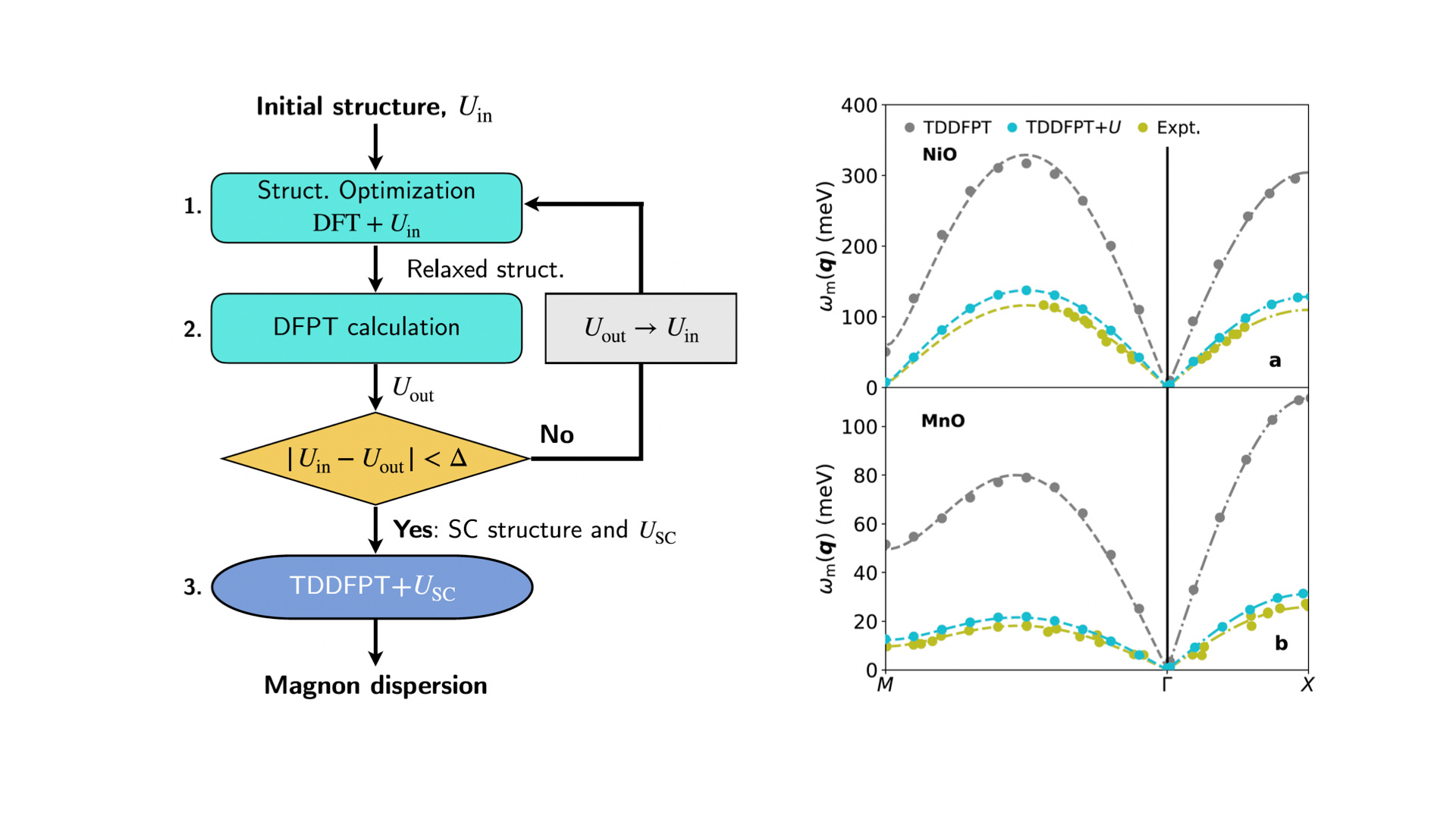
What makes our work unique is the incorporation of Hubbard U corrections computed entirely from first principles, using a DFPT-based linear-response method. These U values are not arbitrarily chosen—they emerge from a physically motivated condition known as piecewise linearity and are obtained via an iterative, self-consistent protocol that also simultaneously optimizes the material’s crystal structure. This makes the entire workflow consistent and predictive, removing a major empirical bottleneck in previous studies.
In practical terms, this means that the magnetic ground state, the electron interactions, the crystal geometry, and the spin excitations are all treated on the same rigorous theoretical footing.
Goldstone’s theorem
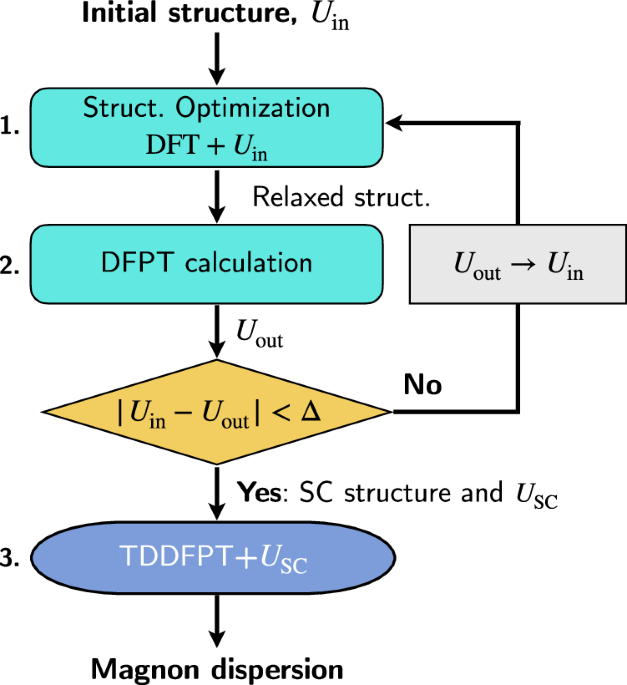
One of the key theoretical challenges in TDDFT-based approaches is the Goldstone condition—the requirement that a system with continuous spin-rotational symmetry should exhibit a magnon mode with zero energy at zero momentum (i.e., a gapless excitation). Many state-of-the-art implementations violate this condition, requiring empirical corrections or sum rule enforcement. Not ours. Our method satisfies Goldstone’s theorem without any ad hoc modifications.
Why Liouville-Lanczos approach?
Among the various technical implementations of TDDFT (Dyson, Sternheimer, etc.), we chose the Liouville-Lanczos (LL) scheme for its exceptional balance between computational efficiency and physical insight. A standout advantage is its ability to compute the frequency-dependent spin susceptibility over a broad spectral range with a single, compact calculation—ideal for systems where the magnon spectrum is unknown or difficult to predict. Moreover, the LL framework avoids the need for explicitly computing empty electronic states—a notorious bottleneck in many-body perturbation theory—and is highly scalable, making it well-suited for high-throughput studies.
Case studies: NiO and MnO
To demonstrate the power of our approach, we applied it to two prototypical Mott insulators: Nickel oxide (NiO) and Manganese oxide (MnO). These materials are classic testbeds for magnetic excitation theories. Our results show remarkable agreement with inelastic neutron scattering data, capturing both the energy and momentum dependence of the magnon spectra with high accuracy. We also show that the subtle rhombohedral structural distortions observed in these materials—often neglected in spin wave models—play a critical role in determining the detailed features of the magnon dispersion. These distortions are correctly captured in our method thanks to the self-consistent optimization of the structure along with the Hubbard corrections.
Looking ahead: A versatile platform
While our current study focuses on prototypical transition-metal oxides, the methodology is broadly applicable to a wide range of magnetic systems, including:
-
Altermagnets, where chiral magnon modes may emerge due to broken spin symmetries.
-
Low-dimensional magnets, such as 2D materials and heterostructures, where experimental data is sparse but theoretical modeling is urgently needed.
The ability to compute accurate magnon spectra without reliance on fitting parameters, spin models, or empirical tweaks positions this method as a next-generation tool for materials discovery and design in magnetism and beyond.
Learn more
To dive deeper into the theory, methodology, and results, check out our full paper in npj Computational Materials:
🔗 https://www.nature.com/articles/s41524-025-01570-0
For a behind-the-scenes perspective, don’t miss the NCCR-MARVEL highlight that explores the motivation and implications of our work:
🔗 https://nccr-marvel.ch/highlights/magnons-transition-metals
Follow the Topic
-
npj Computational Materials
This journal publishes high-quality research papers that apply computational approaches for the design of new materials, and for enhancing our understanding of existing ones.

Related Collections
With Collections, you can get published faster and increase your visibility.
Altermagnetic Materials: Theory, Simulation, and Renewed Perspectives
Publishing Model: Open Access
Deadline: Feb 28, 2027
Recent Advances in Active Matter
Publishing Model: Open Access
Deadline: Sep 01, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in