Mistakes happen, catch them if you can.
Published in Microbiology


In this Behind the Paper post, we look at how we identified and corrected an error while publishing our paper, A framework for assessing 16S rRNA marker-gene survey data analysis methods using mixtures (Olson et al., 2020). During peer-review, we discovered an error in our sample labels when generating a figure requested by a reviewer. Diligent documentation, developing our analysis in a reproducible manner, and the peer review process allowed us to quickly identify, verify, and correct the error. We hope that the lessons learned here will help other scientists prevent, identify, and correct similar errors.
We described our initial mixture and sequencing experimental design in a study protocol document. We developed the initial protocol to minimize the impact of potential sample cross-contamination and propagation of pipetting biases (Fig. 1A). However, the execution of the protocol was not practical. We revised the protocol based on feedback from the co-authors performing the laboratory experiments. Unfortunately, the sample sheet was not updated to reflect these changes. Due to our collaborator’s diligent documentation of the laboratory procedure, we have photographic evidence of the actual sample layout (Fig 1B).
A reviewer requested a PCA analysis of all the samples and titrations to show that the titrations were behaving as expected (Fig. 2A). We generated similar figures during our initial exploratory data analysis but only on individual samples and the expected titration trends were observed. When we plotted the titration series for all five individuals on a single plot, we identified an error in our sample sheet. The plot revealed that the unmixed post-exposure samples were incorrectly grouping for four of the five individuals. From the plot, we hypothesized that we assigned the unmixed post-exposure samples to the wrong individuals. Our hypothesized sample IDs were consistent with the plate layout photo taken in the lab. (Fig. 1B). We corrected the sample IDs and re-generated the PCA analysis, resulting in the expected trend with samples grouping by individual and titration (Fig. 2B). By developing our analysis pipeline in a reproducible manner under version control we quickly reproduced the study results and evaluated the impact of the error on the study conclusions.
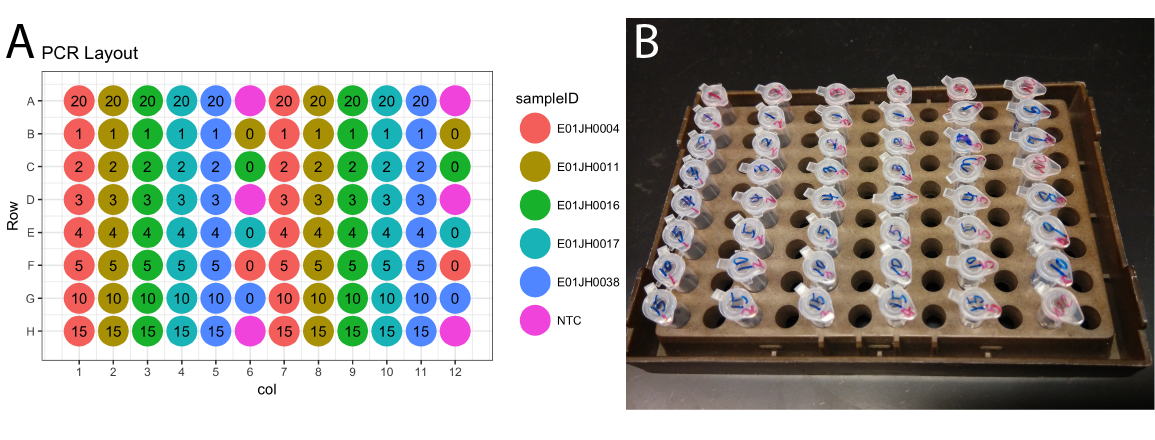
Figure 1: Titration series (A) experimental design plate layout and (B) image of the titration series plate layout for the titration series taken after sample mixing and prior to the initial 16S rRNA PCR.
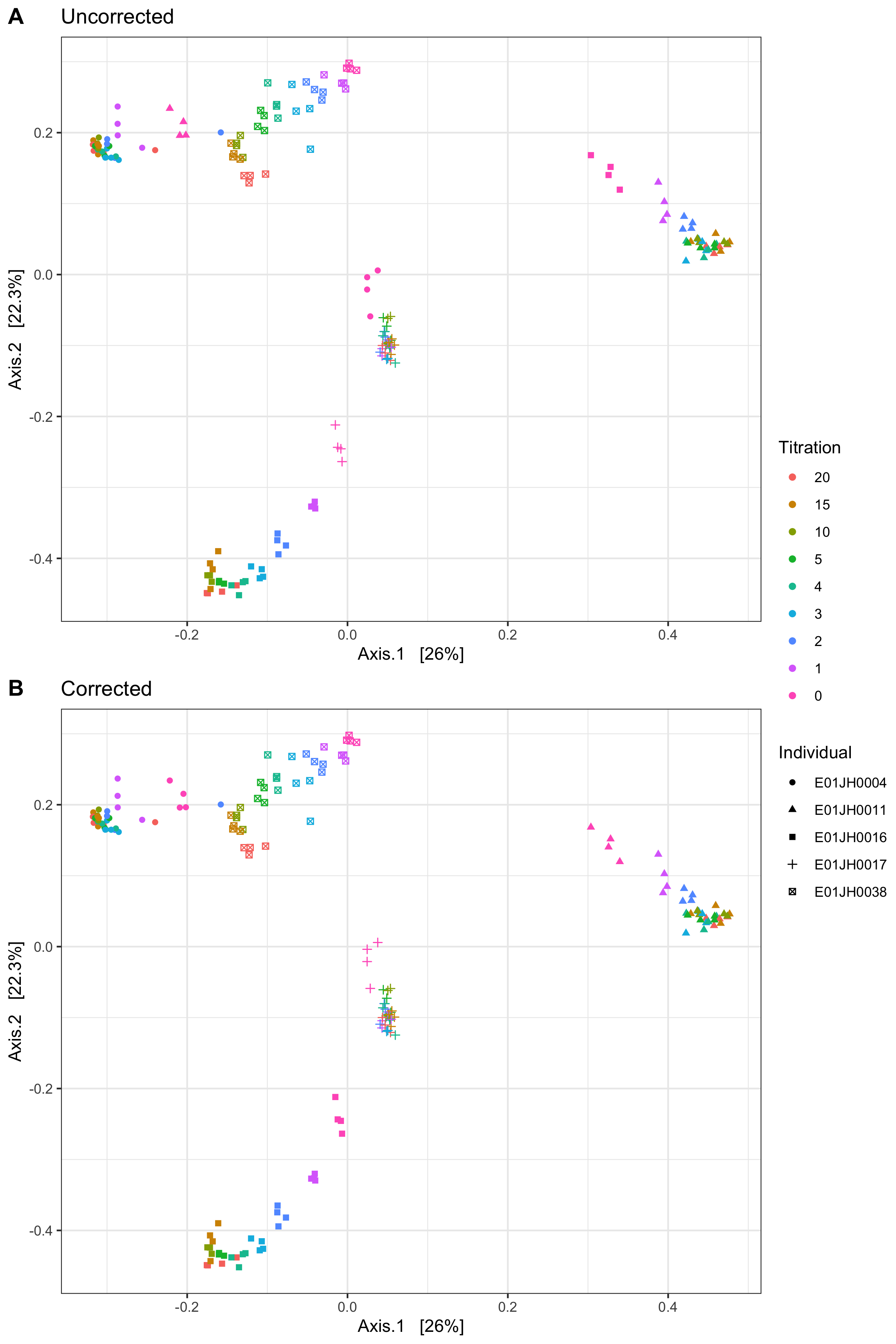
Figure 2: PCA of benchmarking dataset with color indicating titration and shape is individual or subject for the (A) uncorrected sample labels and (B) corrected sample labels. Before sample label correction the unmixed pre-treatment samples, titration 20, are grouped with the incorrect individual for four of the five individuals, E01JH0038 was not effected by the sample sheet error. Whereas, after sample correction the pre-treatment samples were correctly grouped.
It is our hope that documenting and reporting this error will help prevent other researchers from making these same mistakes. While the peer-reviewer process succeeded in identifying the error prior to publication, diligent documentation and a reproducible analysis pipeline allowed us to quickly correct the error and re-run the full analysis pipeline. Let this experience serve as a reminder that mistakes happen in science, and so, it is important to consider preventative measures as a part of any initial study design, perform analyses like PCA that can identify potential errors, and include sample label verification in the protocol.
Reference
Olson, N.D., Kumar, M.S., Li, S. et al. A framework for assessing 16S rRNA marker-gene survey data analysis methods using mixtures.. Microbiome 8, 35 (2020). https://doi.org/10.1186/s40168...
Follow the Topic
-
Microbiome
This journal hopes to integrate researchers with common scientific objectives across a broad cross-section of sub-disciplines within microbial ecology. It covers studies of microbiomes colonizing humans, animals, plants or the environment, both built and natural or manipulated, as in agriculture.

Related Collections
With Collections, you can get published faster and increase your visibility.
Oncobiome
This collection of papers delves into the burgeoning field of oncobiome research, exploring the intricate relationship between cancer and the microbiome. The oncobiome encompasses the diverse microbial communities residing in and on the human body, which influence cancer development, progression, and treatment responses. By examining these interactions, our aim is to unravel the complex mechanisms through which the microbiome impacts oncogenesis and therapeutic outcomes.
This compilation highlights cutting-edge research, offering insights into potential diagnostic markers and novel therapeutic strategies, thereby advancing our understanding of cancer biology and paving the way for innovative, microbiome-targeted cancer treatments.
This is a cross-journal collection between:
Experimental Hematology and Oncology
Articles will undergo the standard peer-review process of the journal to which they are submitted and are subject to either the BMC editorial policies or those of BJC Reports. Articles will be added to the Collection as they are published. The Editors have no competing interests with the submissions which they handle through the peer review process. The peer review of any submissions for which the Editors have competing interests is handled by another Editorial Board Member who has no competing interests.
Publishing Model: Open Access
Deadline: Ongoing
Animal Gut Nutrition and Greenhouse Gas Mitigation
Animal Microbiome, Journal of Animal Science and Biotechnology and Microbiome call for submissions to the collection on Animal Gut Nutrition and Greenhouse Gas Mitigation.
Efforts to reduce greenhouse gas emissions from livestock systems increasingly hinge on innovations in animal gut nutrition. The dynamic relationship between the gut microbiome and nutrient utilization plays a pivotal role in shaping methane output, feed efficiency, and overall sustainability. Advances in microbial ecology—particularly in understanding the role of gut microbiome in nutrient metabolism—are opening new pathways for mitigating emissions while enhancing productivity. These developments support the implementation of climate-smart agricultural strategies to address climate change and its impacts.
Looking ahead, continued research in this field has the potential to yield innovative solutions such as targeted probiotic supplementation, which could further optimize gut function and enhance nutrient absorption. These advancements may lead to reduced greenhouse gas emissions while improving animal health and productivity. By deepening our understanding of the animal gut microbiome, we can contribute significantly to sustainable agricultural practices that benefit both the environment and food security.
We invite researchers to contribute to this special Collection on Animal Gut Nutrition and Greenhouse Gas Mitigation. Topics of interest include but are not limited to:
- Animal Gut Microbiome and Feed Efficiency
- Greenhouse Gas Mitigation Strategies
- Rumen Fermentation Dynamics
- Nutrient Utilization in Livestock
- Probiotic Supplementation Effects
- Sustainable Livestock Production Practices
- Climate-Smart Agriculture Innovations
This Collection supports and amplifies research related to SDG 13, Climate action.
All submissions in this collection undergo the relevant journal’s standard peer review process. Similarly, all manuscripts authored by a Guest Editor(s) will be handled by the Editor-in-Chief of the relevant journal. As an open access publication, participating journals levy an article processing fee (Animal Microbiome fees, Journal of Animal Science and Biotechnology fees, Microbiome fees). We recognize that many key stakeholders may not have access to such resources and are committed to supporting participation in this issue wherever resources are a barrier. For more information about what support may be available, please visit OA funding and support, or email OAfundingpolicy@springernature.com or the Editor-in-Chief of the journal where the article is being submitted.
Publishing Model: Open Access
Deadline: Sep 04, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in