Mitigating the impact of rogue genes in phylogenomic studies
Published in Ecology & Evolution

The advent of genomics and the ever increasing amount of new DNA sequence data generated have given a tremendous boost to phylogenetics, the study of the evolutionary relationships among organisms and their genes, such that we can now seriously contemplate sequencing the genomes of all living organisms. And with the genomes of all living organisms at hand, reconstructing the entire tree of life seems no longer a pipe dream but a realistic and attainable goal.
But how accurate will this genome-based tree of life be? While the vast majority of the tree's branches will likely be robust to different analytical approaches and consistent with our current knowledge of life's history, there is reason to expect that resolution of a few of its branches will prove challenging. Consider, for example, the question whether the sister group to the rest of animals are sponges (also known as poriferans) or comb jellies (also known as ctenophores), which has been vigorously debated for more than a decade now. Although genomic data have greatly helped in reconstructing a more robust tree of animals, they have so far been unable to settle the sponge / comb jelly debate.
In a 2017 study in Nature Ecology & Evolution, we examined the phylogenetic signal for challenging-to-resolve relationships in animal, plant, and fungal phylogenies constructed using genome-scale data. One of our most striking findings was that some of these contentious phylogenetic relationships rested on the phylogenetic signal contributed by a single gene. For example, in a phylogeny of budding yeasts inferred from analyses of a data matrix containing 1,233 genes from 86 species, we noticed that the placement of the fungal family Ascoideaceae, which was represented by the genome of the species Ascoidea rubuscens, was dependent on a single gene with an unusually strong phylogenetic signal. Inclusion of the gene in the 1,233-gene, 86-species data matrix, which goes by the name DPM1 in the baker's yeast Saccharomyces cerevisiae, yielded this topology:
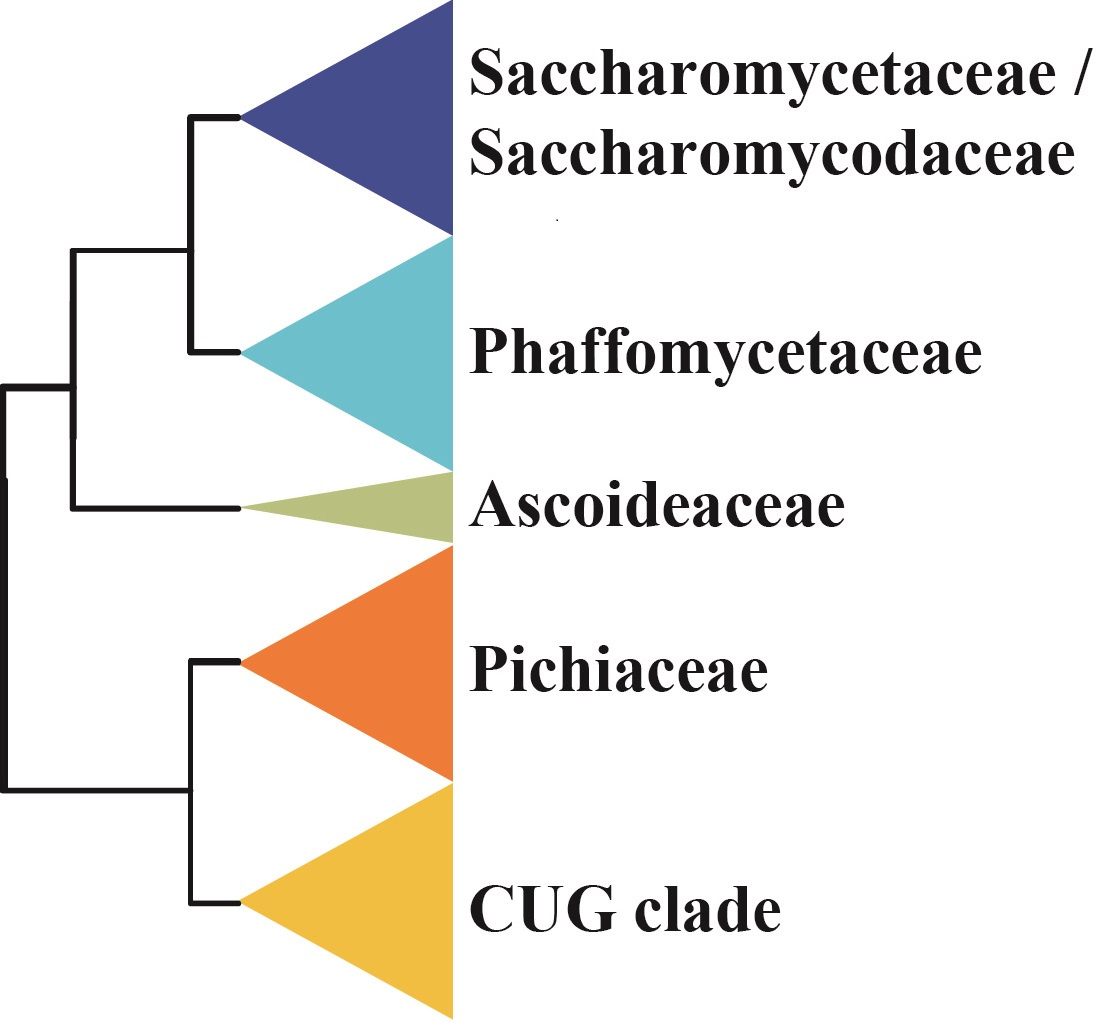
In contrast, removal of DPM1 from the data matrix and analysis of the remaining 1,232 genes yielded this topology:
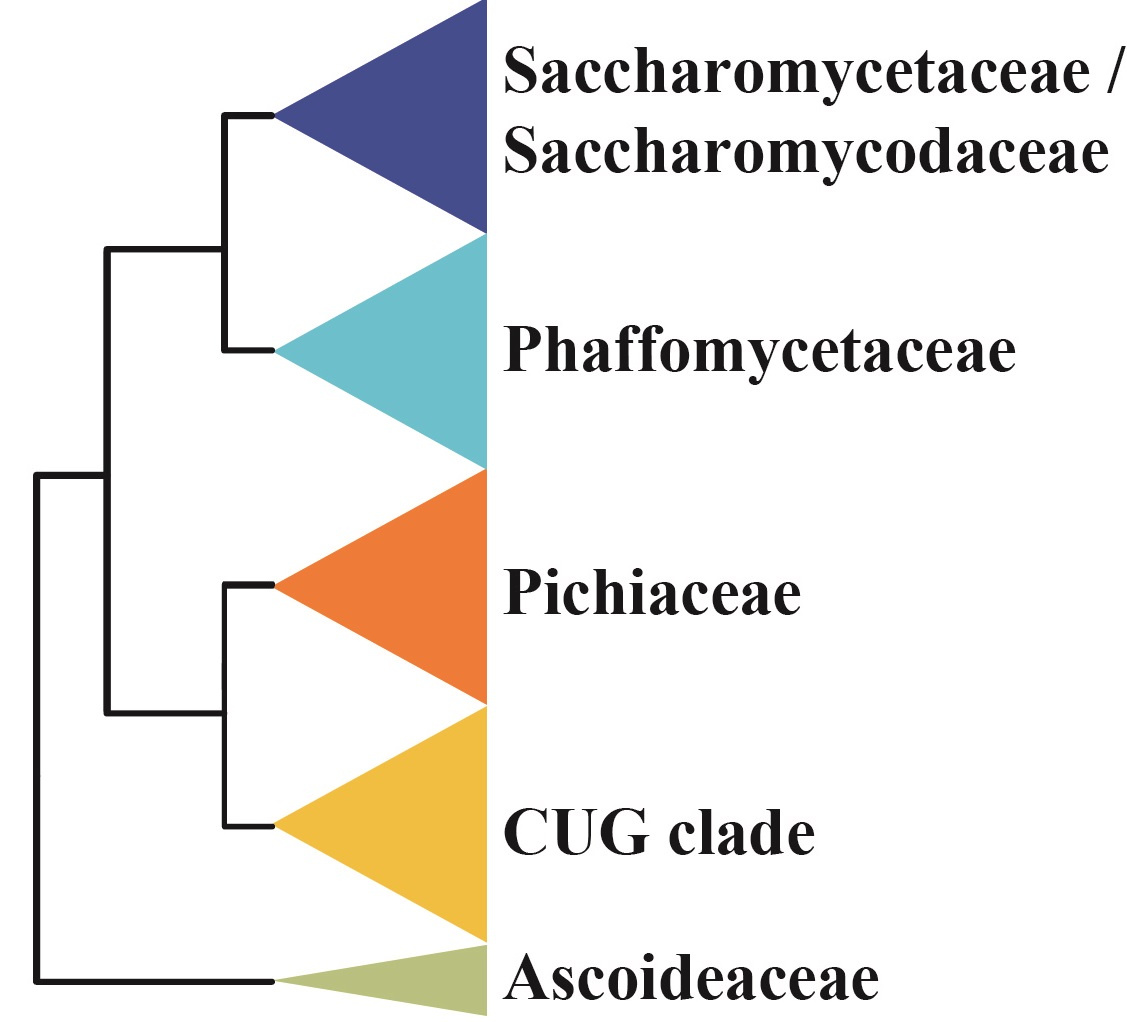
In a study that just appeared online in Cell, we report the analysis of the genomes of 332 budding yeast species, including 220 new ones, that capture nearly a third of known yeast biodiversity. This is work that was done together with Chris Todd Hittinger's lab at the University of Wisconsin-Madison, the late pioneer of budding yeast taxonomy Cletus P. Kurtzman, and several collaborators around the world. But the real drivers of this work were four amazingly talented postdocs: Xing-Xing Shen in my group, Dana Opulente and Jacek Kominek in Chris' lab, and Xiaofan Zhou, a former postdoc in my group who now runs his own lab at Southern China Agricultural University. To put their work in perspective, the largest comparative genomic study in budding yeasts to date is the study by Riley and co-workers (PNAS, 2016), which reported 16 new genomes, whereas the largest comparative genomic study in eukaryotes is the bird tree of life study by Jarvis and co-workers (Science, 2014) that reported 45 new genomes (a study that just appeared in Nature Genetics also reported 45 new genomes of animal parasites; Coghlan et al., 2018).
Although our new study's emphasis is on understanding the evolution of genes and traits involved in metabolism, one of the key achievements of our work is the generation of a genome-scale phylogeny and timetree that captures the diversity of budding yeasts (our analyses included genomes from 79 of the 92 recognized budding yeast genera!). Here's an image of it where species names have been removed and each lineage is shown with a different color (Ascoideaceae is part of the CUG-Ser2 clade in light green around the 5 hour mark):
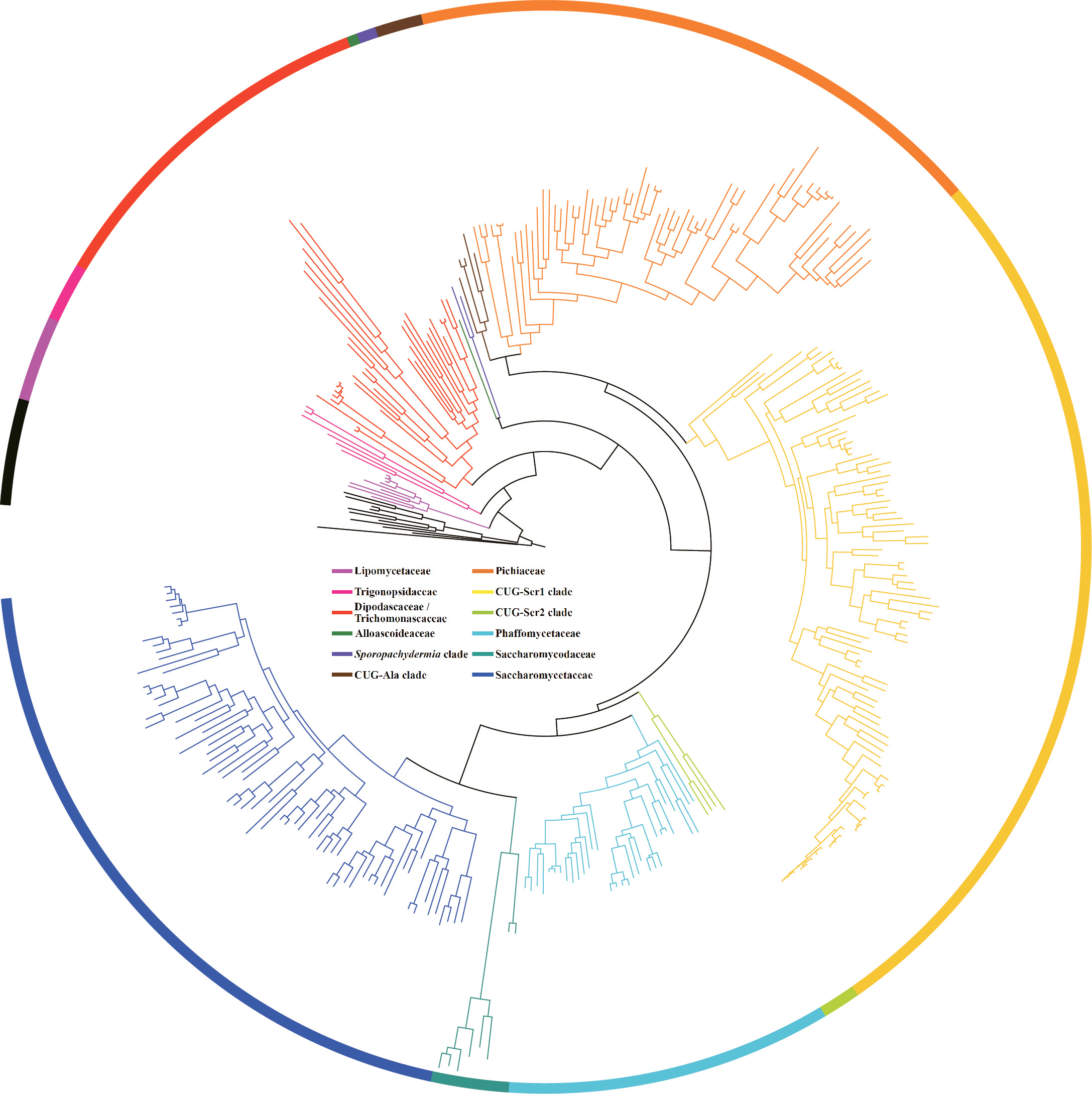
Painstaking examination, both via different types of analyses as well as by analyses of different subsets of genes, of the robustness of this new budding yeast phylogeny revealed that ~10% (32 / 331) of branches show conflict between analyses. Interestingly, this level of conflict is lower than ~13% (11 / 85), the level we observed in our 2016 analyses of the 86-species budding yeast phylogeny.
But what about the placement of the family Ascoideaceae and the CUG-Ser2 clade in the new, expanded budding yeast phylogeny? The addition of the genomes of three additional species appears to have solidified support for this topology:
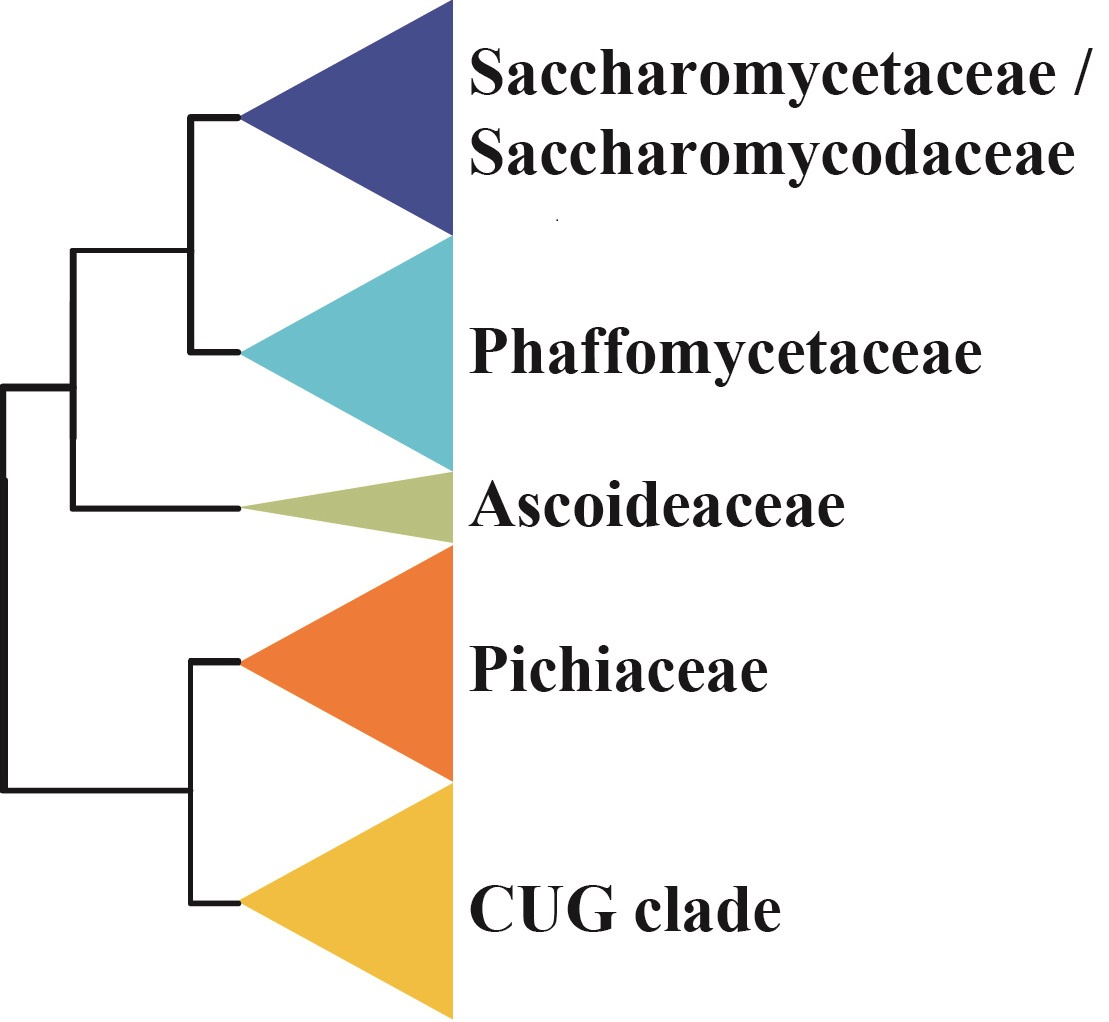
And the DPM1 gene, you may ask? Remarkably, inclusion of the DPM1 sequences from the three new species in the Ascoideaceae / CUG-Ser2 clade appears to have dramatically reduced the gene's phylogenetic signal. Interestingly, when we exclude these three species from the data matrix, DPM1 regains its unusually strong phylogenetic signal:
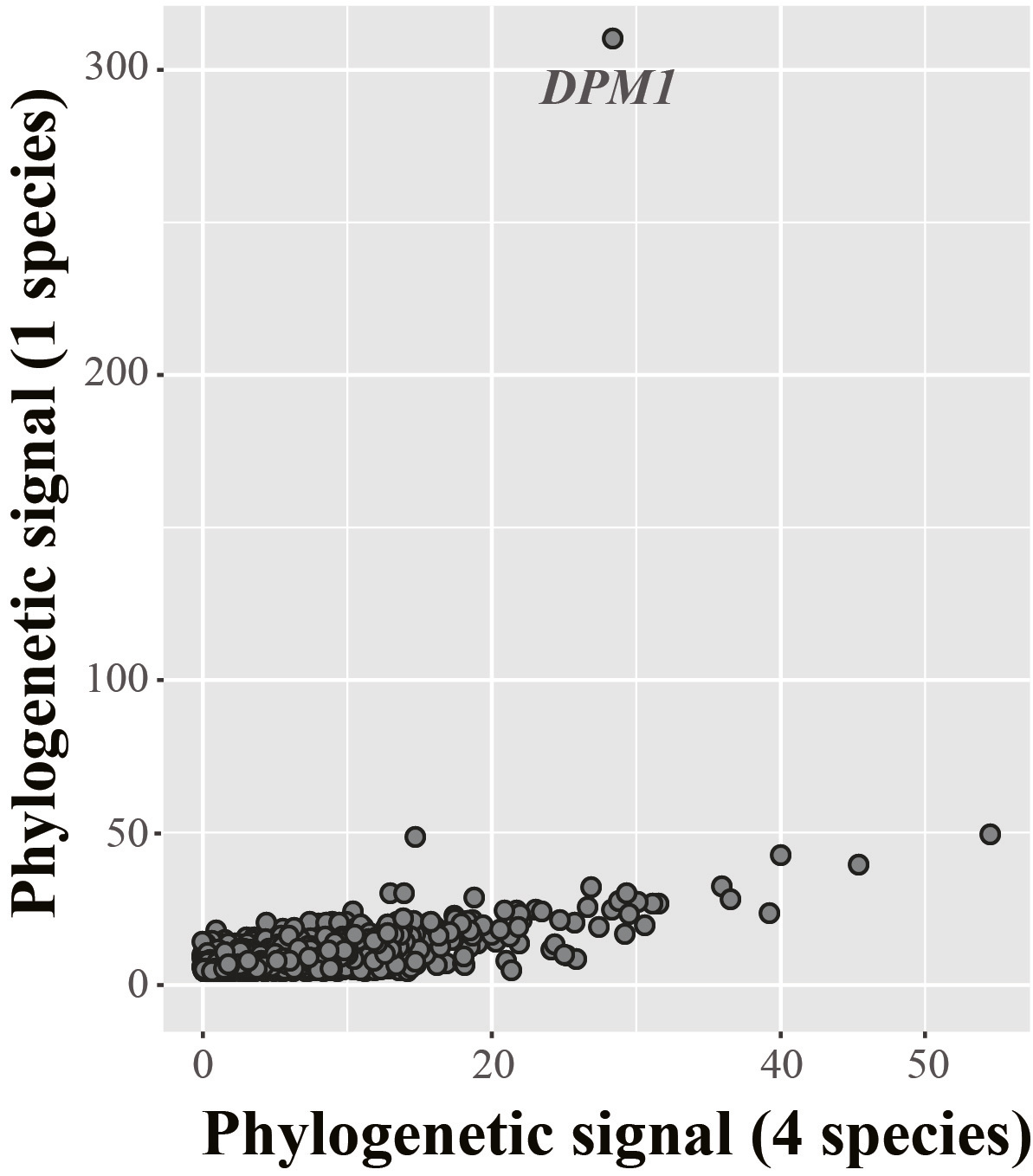
This behavior of the DPM1 gene is consistent with the explanation that the model of sequence evolution used to describe the gene's evolution shows a poor fit to the actual data at hand. Increasing the number of DPM1 sequences from one to four for just the lineage in question appears sufficient to improve the model's fit to the data, ameliorating the gene's undue impact on the placement of this lineage on the tree of budding yeasts.
These results are consistent with simulation studies showing that adding species can dramatically increase phylogenetic accuracy. This is good news for efforts to assemble life's family tree from genomic data. If current controversies in phylogenetics are any indication, robustly inferring life's entire family tree will require all the help that we can get!

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in