Why are some evolutionary relationships so controversial?
Published in Ecology & Evolution

In his magnum opus, Darwin prophesized that “we have to discover and trace the many diverging lines of descent in our natural genealogies, by characters of any kind which have long been inherited”. In the last decade or so, the genomics revolution dramatically accelerated the generation of genome-scale DNA data for inferring life’s genealogy, the so-called tree of life, ushering us in the era of phylogenomics. Not surprisingly, as the size of our data and the sophistication of our phylogenetic algorithms increased, the resolution of the tree of life increased too.
But some relationships have refused to yield to the new approach, with different studies producing strongly contradictory answers. For example, are sponges the sister lineage to all other animals? Or are comb jellies (aka ctenophores)? Opinions differ and ever-larger analyses have failed to generate consensus 1–4. Why is that so? And how do we move forward?
To shed light on these questions, Xing-Xing Shen, a talented postdoc in my lab at Vanderbilt University, longtime collaborator Chris Todd Hittinger of the University of Wisconsin-Madison, and I decided to specifically focus our investigations on well-known controversial branches, so we picked 5 from plants, 6 from animals, and 6 from fungi as well as 6 well-established “control” branches, and got going.
To examine the nature of the conflict for these controversial relationships, we harnessed the powerful framework of the maximum likelihood approach to examine the gene- and site-level phylogenetic signal in favor of the two best-supported alternative phylogenetic hypotheses (we call them T1 and T2) for a given controversy (note: we did not come up with this approach; several others have previously used it, to great success 5–7).
Doing so allowed us to precisely quantify the distribution of phylogenetic signal for T1 and T2, as well as visualize the proportions of sites’ or genes’ support for the controversy (T1 vs T2) surrounding each one of our branches, as shown in this image:
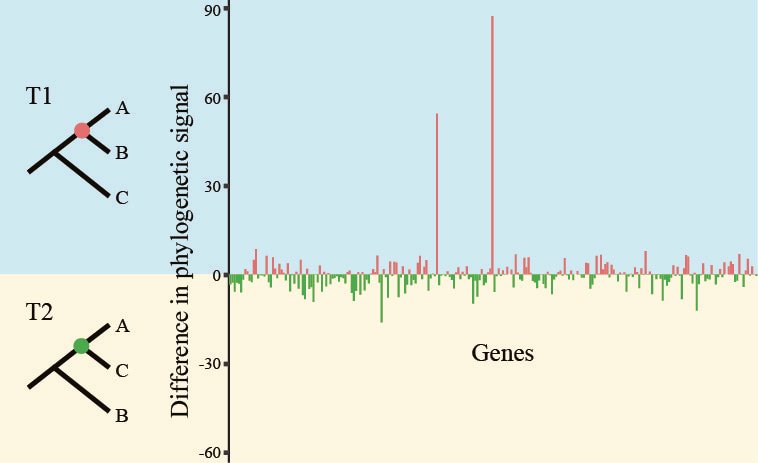
By examining all 17 contentious branches, we noticed that, in half a dozen or so branches, a single or handful of genes displayed very strong signal (note the bars with the very large values in favor of T1 in the image above), arguing that the support in favor of one hypothesis stemmed from a few genes rather than from hundreds of them.
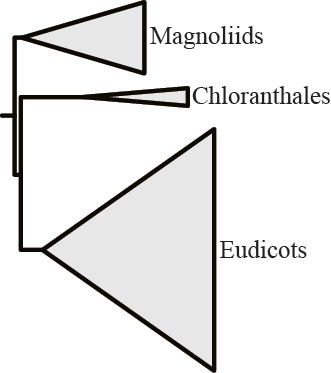
To our great surprise, in 3 branches, removal of the “most strongly opinionated” single gene eliminated support for T1 and boosted support for T2 (needless to say, none of our controls showed this behavior)! One of them concerns relationships among flowering plants, where analysis of the 103-taxon, 620-gene rich data matrix yielded this result:
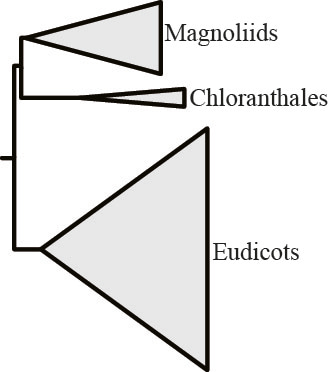
But removal of the most opinionated gene and reanalysis of the now 619-gene rich data matrix, yielded a different result:
What we draw from these experiments is that tiny amounts of data in otherwise very large phylogenomic data matrices exert decisive influence in the resolution of certain contentious branches.
What does this mean? In my opinion, this means that relationships that show this kind of behavior should be considered unresolved. Of course, this is not to say that they will never be resolved; just that current data and methods are equivocal. Resolving them will require more data, different types of data, or more sensitive methods (or all three!).
We also tested whether the support for the 17 contentious relationships was also sensitive to “highly opinionated sites” within genes by examining the effect of removing the site with the strongest phylogenetic signal from every gene. In phylogenomic data matrices containing tens or hundreds of thousands of sites from hundreds of genes removal of a few hundred sites should not mean a thing, yet here we too found that this removal had a huge influence, altering the support in 9/17 contentious branches! Interestingly, a couple of branches, including the plant branch we discussed above as well as relationships at the base of the family tree of modern birds, were susceptible to both the removal of the single gene as well as to the removal of a single site per gene!
But our approach can also augment the support for one hypothesis over another. For example, examination of the evolutionary placement of crocodiles strongly supported the hypothesis that they are the sister group to birds over the second best alternative (that they are the sister group to turtles)8,9:
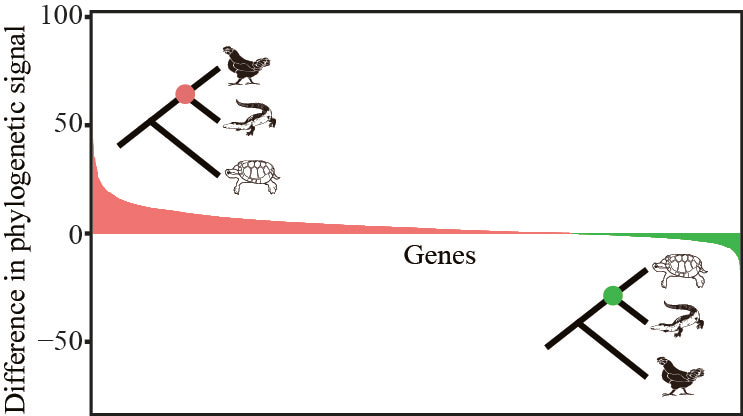
In this image, we’ve ranked the genes from those most highly in favor of crocodiles + birds (shown in red) to those most highly in favor of crocodiles + turtles (in green). Note that the area under the red curve is much larger than the green one, arguing strongly in favor of crocodiles + birds. Interestingly, a recent study using Bayesian inference to evaluate the relative support for the two alternatives came up with the same answer 10.
To drive this important point home, our final experiment examined the support for what is turning out to be the “mother of all phylogenetic controversies”: the three alternative, hypotheses regarding the sister lineage to the rest of animals.
Is it the sponges?
.jpg)
Is it the comb jellies?

Or is both? Our investigation of 8 published phylogenomic data sets showed that the “comb jellies-first” hypothesis always had the highest proportions of supporting genes and sites, and was the most robustly supported. For example, here are the percentages of genes favoring each of the tree alternatives for one of the data matrices we analyzed (Whelan_Dataset16_Choanoflagellata):
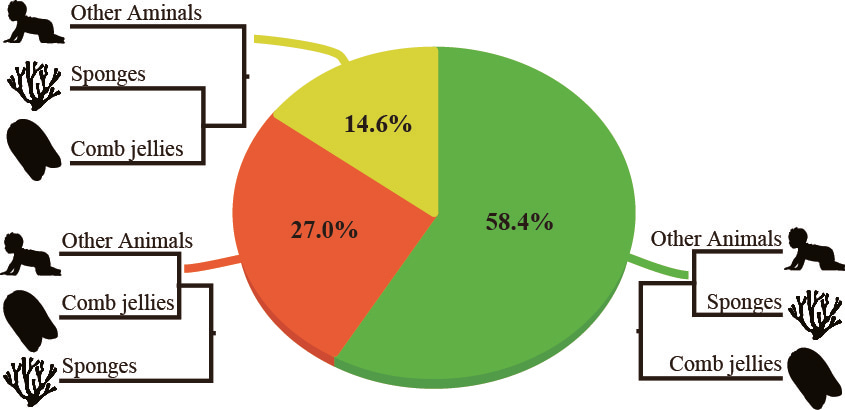
While I am confident that our analysis will not be the last word on the “fight over animal origins” (a phylogenomic study published just last week weighs in favor of sponges 4,11), by quantifying the support for all three alternative hypotheses, our approach illuminates the controversy and shows a path toward its resolution.
The paper in Nature Ecology & Evolution is here: http://go.nature.com/2oueCvB
References
1. Dunn, C. W. et al. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature 452, 745–749 (2008).
2. Whelan, N., Kocot, K. M., Moroz, L. L. & Halanych, K. M. Error, signal, and the placement of Ctenophora sister to all other animals. Proc. Natl Acad. Sci. USA 112, 5773–5778 (2015).
3. Ryan, J. F. et al. The genome of the ctenophore Mnemiopsis leidyi and its implications for cell type evolution. Science 342, 1242592 (2013).
4. Simion, P. et al. A Large and Consistent Phylogenomic Dataset Supports Sponges as the Sister Group to All Other Animals. Curr. Biol. 27, 958–967 (2017).
5. Castoe, T. A. et al. Evidence for an ancient adaptive episode of convergent molecular evolution. Proc. Natl Acad. Sci. USA 106, 8986–8991 (2009).
6. Shavit Grievink, L., Penny, D. & Holland, B. R. Missing data and influential sites: choice of sites for phylogenetic analysis can be as important as taxon sampling and model choice. Genome Biol. Evol. 5, 681–687 (2013).
7. Kimball, R. T., Wang, N., Heimer-McGinn, V., Ferguson, C. & Braun, E. L. Identifying localized biases in large datasets: a case study using the avian tree of life. Mol. Phylogenet. Evol. 69, 1021–1032 (2013).
8. Chiari, Y., Cahais, V., Galtier, N. & Delsuc, F. Phylogenomic analyses support the position of turtles as the sister group of birds and crocodiles (Archosauria). BMC Biol. 10, 65 (2012).
9. Shen, X.-X., Liang, D., Wen, J.-Z. & Zhang, P. Multiple genome alignments facilitate development of NPCL markers: a case study of tetrapod phylogeny focusing on the position of turtles. Mol. Biol. Evol. 28, 3237–52 (2011).
10. Brown, J. M. & Thomson, R. C. Bayes factors unmask highly variable information content, bias, and extreme influence in phylogenomic analyses. Syst. Biol. (2016). doi:10.1093/sysbio/syw101
11. Maxmen, A. Big data renews fight over animal origins. Nature (2017). doi:10.1038/nature.2017.21703
Follow the Topic
-
Nature Ecology & Evolution
This journal is interested in the full spectrum of ecological and evolutionary biology, encompassing approaches at the molecular, organismal, population, community and ecosystem levels, as well as relevant parts of the social sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Understanding species redistributions under global climate change
Publishing Model: Hybrid
Deadline: Dec 31, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in