Mitochondria: The powerhouse of somatic mutation
Published in Ecology & Evolution and Genetics & Genomics
Probing somatic evolution of the mitochondrial genome
Our genetic information is housed in two genomes: the nuclear and mitochondrial genomes (mt-genome). The mt-genome encodes for 13 vital proteins. However, more than 1,000 genes encoded on the nuclear genome are necessary for mitochondrial assembly and function. This tight-knit coordination requires the two genomes to be on the same page and to co-evolve. Unique to the mt-genome is that it is maternally inherited, whereas the nuclear genome is inherited from both parents. This characteristic can lead to differences in ancestry between the nuclear and mt-genome. Studies across a wide range of hybrid species have showcased that mismatches between mitochondrial and nuclear ancestries can result in reductions in fitness. In 2019 Patrick Chinnery’s group demonstrated that this difference in ancestry could bias the mutations transmitted to the following generation (Wei et al. 2019), favoring mito-nuclear matching. Together a plethora of studies at both population and molecular scales have clearly demonstrated the functional importance of nuclear-mitochondrial interactions.
The mt-genome presents an incredibly complex system to study rapid changes in the genome. The mt-genome is a high copy number genome, with hundreds to thousands of genomes within a single cell. On top of this, the mt-genome has a 10- to 100- fold higher somatic mutation rate than the nuclear genome. Thus, mt-genomes are a dynamic subcellular population with the ability to change within an individuals’ lifespan.
Within the last decade, advancements in sequencing technologies have allowed us to uncover and catalog mutations in the mt-genome at an unprecedented resolution. While next generation sequencing approaches have given us the power to catalog population genetic variation in genomes, they have error rates of ~1%. This means that we cannot confidently recognize a mutation present at a frequency <1% in the population of mt-genomes present in an individual. Duplex sequencing, an ultra-sensitive sequencing technique, uses unique molecular identifiers to tag the duplex mtDNA molecule. This allows us to sequence DNA at unprecedented accuracy, with an error rate of <10-7. With this sequencing tool, we can understand the complete spectrum of mutations aggregating on mt-genomes and are not limited to high-frequency mutations.
Within the last few years, various groups have used Duplex Sequencing in efforts to pinpoint somatic mutations. These works have confirmed that mt-genomes exhibit increased somatic mutation rates, that replication-associated errors are the primary source of mutation, and that there exists tissue-specific differences in somatic mutation rates (to add to the complexity!). In our work, we explored the impact of mitochondrial ancestry and nuclear-mitochondrial interactions on the somatic evolution of the mt-genome.
A deep resource of mitochondrial somatic mutation and evolution
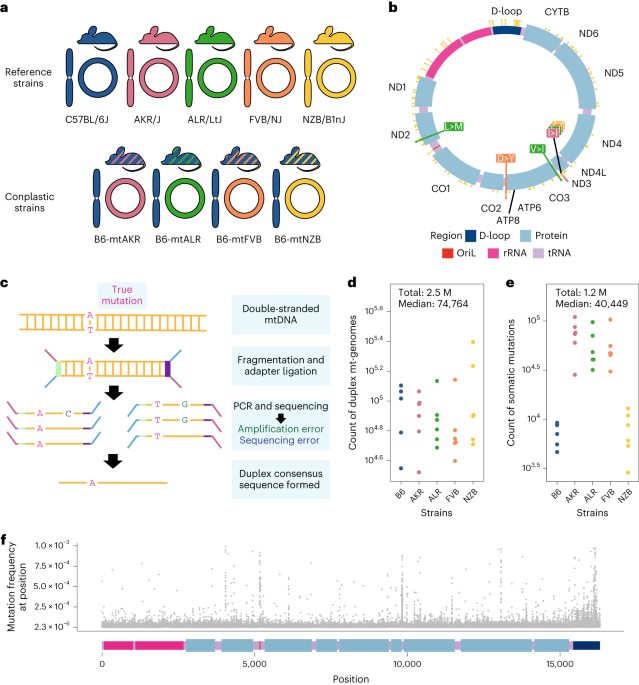
We employed a panel of four mouse strains that are identical in their nuclear genomes (B6) but differ at fixed sites in their mt-genomes (so called “conplastic” mouse strains). As a control, we used wildtype B6 mice that have “matched” B6 nuclear and mitochondrial ancestry. By fixing the nuclear ancestry across our experiments, we were able to study how discordance between nuclear and mitochondrial genomes impacted mutation. Importantly, two of the conplastic mouse strains differ from the B6 mt-haplotype by just 1-2 nonsynonymous variants while one strain differs by 91 variants across the entire mt-genome. Thus, the conplastic mice have varying degrees of mito-nuclear ancestral mismatching. Using duplex sequencing we profiled ~2.5 million mt-genomes cataloging ~1.2 million low frequency inherited and somatic mutations. This resource allowed us to accurately capture mutations with a frequency as low as of 4x10-6, or a mutation present in 1 out of ~200,000 mt-genomes (Serrano et al. 2024).
Hotspots of mutation across vast evolutionary distances

Fig. 1: Comparison of mutational hotspots in the OriL in mice to macaques. Positions with a mutation frequency > 1x10-3 are in magenta, while positions that undergo mutations are denoted in light blue. In the macaque OriL diagram, variant hotspots are denoted in light blue. Stars represent mouse strains that have mutations present at a given position (B6 (blue), AKR (pink), ALR (green), FVB (orange), and NZB (yellow)).
We began our exploration by characterizing the frequency of mutations in the mt-genome and identifying regions that were prone to mutation, or mutational hotspots. We identified three hotspots, including the Origin of Replication of the Light Strand (OriL), which was recently identified as a hotspot in mtDNA in macaque liver (Arbeithuber et al. 2022). Notably, the mouse and macaque mtDNA OriL sequences are vastly different, however they share a conserved stem loop structure. Remarkably, mutations occurred in the same locations of this stem loop between the two species providing insights into the mechanistic basis of these mutations.
The mitochondrial somatic mutational spectrum differs among species
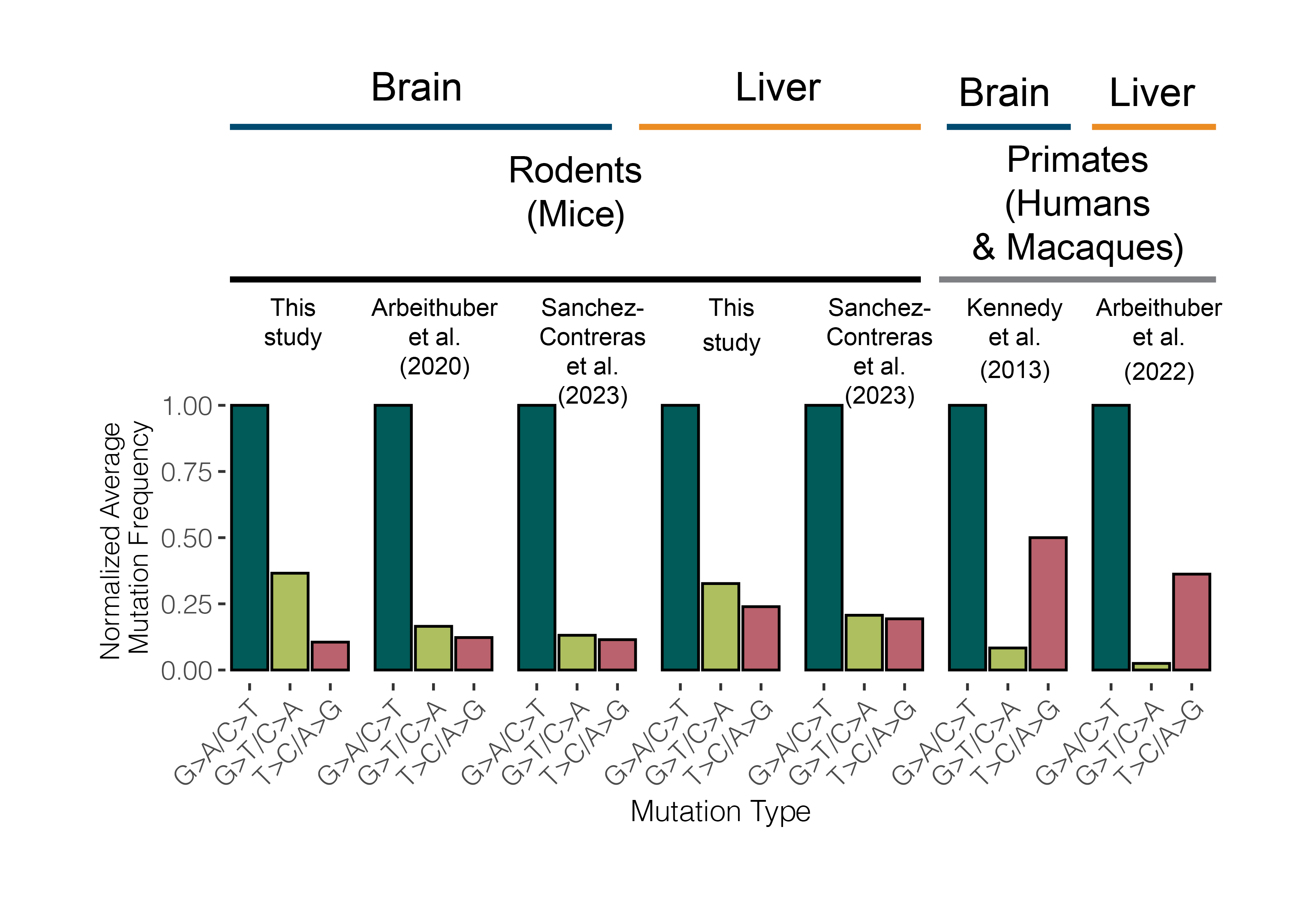
Mutations serve as fingerprints of the molecular processes that damage mtDNA. Commonly known fingerprints are G>A and T>C mutations, which reflect mtDNA replication-associated damage, and G>T mutations, which arise from metabolism induced reactive oxygen species. In 2013, a study of somatic mitochondrial mutation in the human brain found the two most common mutations to be G>A and T>C, i.e. replication associated (Kennedy et al. 2013). However, in our data we found the reactive oxygen induced G>T mutations were present at a higher frequency than T>C mutations. To investigate this discrepancy, we gathered duplex mtDNA sequencing data across macaques, mice, and humans to compare the mutational spectra of primates to rodents. We found that rodents exhibit a surfeit of G>T mutations compared to humans and macaques. These results are consistent with recent work profiling somatic mutation in the nuclear genome (Cagan et al. 2022). Together these results suggest species-specific differences in mutation that potentially correlate with differences in other life history traits (e.g. body size, lifespan, metabolic rate).
Mitochondrial genomes undergo somatic evolution to match nuclear ancestry
Many hybrid species with mismatched mito-nuclear genomes exhibit reduced fitness. We thus asked if somatic mutations that “re-aligned” mitochondrial and nuclear ancestry were more common than expected, suggesting a somatic selective benefit. In mouse strains with mismatching ancestry we compared the frequency of such “reversion” mutations to the background mutation rate. Reversion mutations were 7- to 100-fold more common than expected in these strains. Furthermore, these mutations by-and-large increased in frequency with age. These results highlight that interaction between the ancestry of nuclear and mitochondrial genomes influences selective trajectories even on very short timescales such as through the lifespan of an individual.
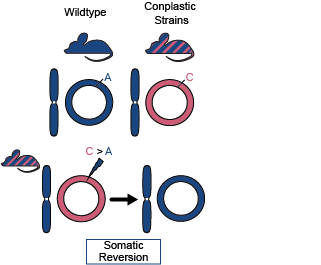
Fig. 3: Schematic of somatic reversion mutations. The wildtype strain (B6) has matching nuclear and mt-genome ancestries (B6 ancestry denoted in blue). Conplastic strains are hybrids with mismatching nuclear and mt-genome ancestry. Haplotype sites are positions in the mt-genome where the conplastic mt-genome differs from the B6 mt-genome. Somatic reversion mutations refer to the reintroduction of B6 ancestral alleles at haplotype sites.
Future work
Our results begin to describe how the mt-genome is shaped at various scales. At the species level, our work demonstrates that mutational processes differ based on life history traits. However, broader comparative analysis across taxa using ultra-sensitive sequencing is needed to fully understand differences in mutational processes across species. At the organismal level, mutation frequencies are tissue-specific with the dominant mutational signature being associated with replication error. However, in order to parse out the roles played by different mutational processes within tissues single cell approaches are needed. Lastly, with respect to the role ancestry plays on mutation, we show that throughout aging there is a preference to re-align the mitochondrial and nuclear ancestries. To further dissect the roles of nuclear and mitochondrial genomes in dictating selective processes, deep somatic profiling of both the nuclear and mitochondrial genomes from species with reciprocal nuclear/mt-genome swaps should be attempted.
References
- Wei, W. et al. Germline selection shapes human mitochondrial DNA diversity. Science 364, (2019).
- Serrano, I. M. et al. Mitochondrial haplotype and mito-nuclear matching drive somatic mutation and selection throughout ageing. Nature Ecology & Evolution 1–14 (2024).
- Arbeithuber, B. et al. Advanced age increases frequencies of de novo mitochondrial mutations in macaque oocytes and somatic tissues. Proc. Natl. Acad. Sci. U. S. A. 119, e2118740119 (2022).
- Kennedy, S. R., Salk, J. J., Schmitt, M. W. & Loeb, L. A. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 9, e1003794 (2013).
- Cagan, A. et al. Somatic mutation rates scale with lifespan across mammals. Nature 604, 517–524 (2022).
Header image inspired by the schematic from FamilyTreeDNA.
Follow the Topic
-
Nature Ecology & Evolution
This journal is interested in the full spectrum of ecological and evolutionary biology, encompassing approaches at the molecular, organismal, population, community and ecosystem levels, as well as relevant parts of the social sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Understanding species redistributions under global climate change
Publishing Model: Hybrid
Deadline: Dec 31, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in