Mitochondrial Dysfunction in Charcot Marie Tooth 2B peripheral neuropathy?
Published in Healthcare & Nursing and Neuroscience

What is happening with mitochondria in Charcot Marie Tooth 2B peripheral neuropathy?
Charcot Marie Tooth (CMT) diseases
CMT peripheral neuropathy is named after three physicians-Jean Martin Charcot, Pierre Marie, Howard Henry Tooth who first described the disease. The disease affects both the peripheral sensory and motor nerves. Genetically, it is extremely complexed.

CMT affects about 2.8 million people worldwide, of all races and ethnic groups. There are more than 80 types of CMT, each caused by a different mutation. The CMT type 2B (CMT2B) is a rare axonal CMT with clinical manifestations of severe and progressive peripheral sensory neuropathy. Currently here is no effective treatment. The onset of CMT2B is as early as 20-30 years old. The main features of CMT2B include:
- Symmetric and severe distal sensory loss, mostly in lower legs and feet
- Progressive distal weakness and muscle atrophy
- Feet deformities
- Ulcerations and infections in feet
- Reduce tendon reflects
- Brain functions are almost never affected
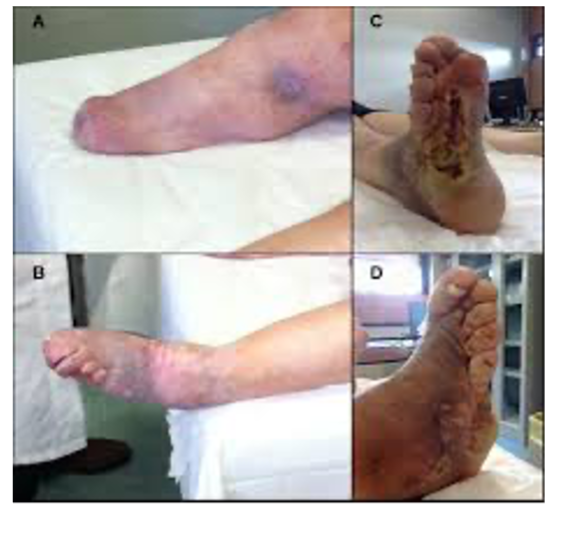
Images are from Manganelli, F. et al. Autonomic nervous system involvement in a
new CMT2B family. J Peripher Nerv Syst 17, 361-364 (2012) 4.
Etiology of CMT2B
CMT2B is inherited by an autosomal dominant manner, caused by 5 missense mutations (p.L129F, p.K157N, p.N161T/I and p.V162M) in the RAB7A gene located on Chromosome 3. Interestingly, a novel RAB7A mutation (p.K126R) was recently reported to induce a sensorimotor CMT2B phenotype5.
What is RAB7A gene?
RAB7A gene is also referred to as RAB7. The RAB7 protein is ubiquitously expressed and plays a pivotal role in endocytic trafficking. In neurons, RAB7 regulates axonal trafficking of neurotrophic factor signaling and mediates lysosomal and autophagic function.
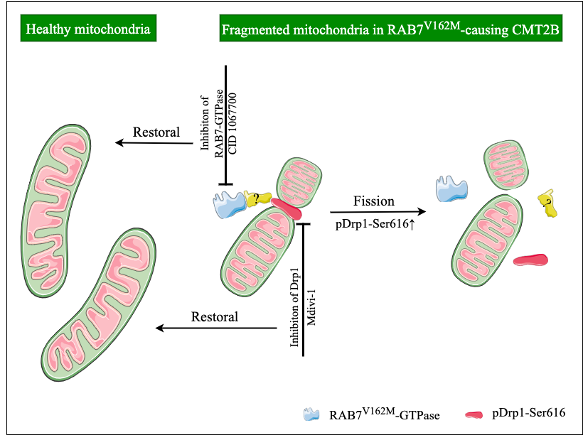
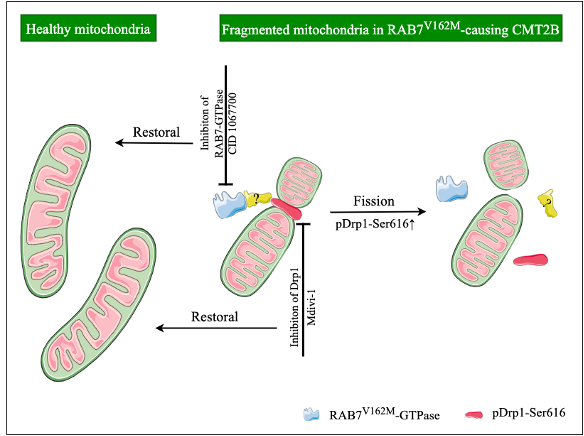
Ongoing research continues to discover more potential roles of RAB7 on mitochondrial structure, motility and function. On one hand, RAB7 regulates mitochondrial fission/fusion process through phosphorylation of dynamin-related protein 1 (pDrp1-Ser616) and the mitochondrial fusion protein MFN2. RAB7 also might affect the crosstalk between mitochondria and lysosomes, in turn to regulate mitochondrial morphology. On the other hand, RAB7 promotes the removal of damaged mitochondria by controlling expansion of the autophagic membrane around damaged mitochondria during mitophagy. Other function of RAB7 is to be involved in the translation of mRNAs encoding mitochondrial proteins at the late endosomal level.
How does CMT2B-RAB7 mutant protein affect mitochondria?
Although the precise pathogenic mechanism(s) of CMT2B by RAB7 mutations is unknown, increasing evidence suggests that mitochondrial structure and function is compromised by Rab7 in CMT2B. define how mitochondria were impacted in CMT2B, we and our collaborators examined CMT2B mouse model and fibroblasts from CMT2B human patients in a recent publication in Communication Biology6.
The diagram was made using Diagram.drawio.The bioicons were downloaded from https://bioicons.com/extensions/.
We first asked if mitochondria were compromised in Rab7V162M-causing CMT2B. So we collected and cultured the skin fibroblasts from human patients, as well as mouse embryonic fibroblasts (MEFs) from a Rab7V162M knockin mouse model. The fibroblasts were then incubated with Mito Tracker and mitochondrial images were captured by live cell imaging. From the analysis and measurement of the size and shape of individual mitochondrion, such as area, perimeter, aspect ratio (maximal:minimal diameter) and the form factor. We found significant mitochondrial fragmentation in human patient fibroblasts and MEFs of CMT2B mouse. By quantifying the network complexity, we found that the mitochondrial fragmentation led to a reduction in the branch numbers, branch length of individual mitochondrion, and branch joints among mitochondria.
Next, we asked what factor(s) contributed to the mitochondrial fragmentation in CMT2B. We examined the pDrp1-Ser616 level in human patient fibroblasts. The data showed a significant increase in the ratio of pDrp1-Ser616/total Drp1 in human CMT2B patient than healthy control. These findings suggest that increased level of Drp1 (pS616) likely contributes to excessive fragmentation in CMT2B.
Pathologically, CMT2B is known to selectively afflict axonal functions of peripheral sensory neurons. To investigate whether or not peripheral sensory neurons had similar changes in mitochondrial morphology as MEFs in our CMT2B mouse model, we cultured dorsal-root-ganglion (DRG) sensory neurons from the CMT2B mouse model. We measured both mitochondrial size and mitochondrial motility in axons. Similar to human fibroblasts and MEFs, DRG sensory neurons showed that mitochondria were significantly fragmented, also mitochondrial network complexity was reduced. Interestingly, axonal mitochondrial moving speeds were increased in the mutant DRG neurons in a gene-dosage dependent manner.
Given that Rab7 is ubiquitously expressed in all cell types and clinically CMT2B patients are not known to suffer from any deficits in their brain functions. we further investigated if mitochondria were impacted in neurons from the central nerve system (CNS). We cultured both hippocampal and cortical neurons from our CMT2B mouse model, followed by measuring mitochondrial size. However, we did not detect significant changes in either hippocampal or cortical primary neurons when we compared CMT2B with WT control samples.
To establish that the RAB7 protein was hyperactivated which, in turns, activates Drp1, leading to excessive fragmentation of mitochondria in CMT2B. We treated the cultured MEFs and DRG neurons with inhibitors of either RAB7 (CID1067700) or Drp1 (Mdivi-1). We found that inhibition of Drp1 or Rab7 fully normalized the mitochondrial deficits in both MEFs and E18 cultured DRG neurons.
Conclusions from our study
We believe that our studies have uncovered, for the first time, a novel mechanism for the pathogenesis of CMT2B, in which the expression of CMT2B-causing RAB7 mutation enhances pDrp1-Ser616 activity to promote mitochondrial fission, that may potentially underlie selective vulnerability of peripheral sensory neurons in CMT2B. These findings will not only contribute significantly to our understanding of the disease but also point to that selectively targeting DRP1 with small molecular inhibitors may represent an effective treatment strategy for the disease.
References:
1 Waraich, M. & Shah, S. The life and work of Jean-Martin Charcot (1825-1893): 'The Napoleon of Neuroses'. J Intensive Care Soc 19, 48-49 (2018). https://doi.org:10.1177/1751143717709420
2 Pearce, J. M. Howard Henry Tooth (1856-1925). J Neurol 247, 3-4 (2000). https://doi.org:10.1007/s004150050002
3 Poirier, J. & Chretien, F. Pierre Marie (1853-1940). J Neurol 247, 983-984 (2000). https://doi.org:10.1007/s004150070062
4 Manganelli, F. et al. Autonomic nervous system involvement in a new CMT2B family. J Peripher Nerv Syst 17, 361-364 (2012). https://doi.org:10.1111/j.1529-8027.2012.00415.x
5. Saveri P, et al. Charcot-Marie-Tooth Type 2B: A New Phenotype Associated with a Novel RAB7A Mutation and Inhibited EGFR Degradation. Cells. 2020 Apr 21;9(4):1028. doi: 10.3390/cells9041028. PMID: 32326241; PMCID: PMC7226405.
6. Gu, Y., Guerra, F., Hu, M. et al. Mitochondria dysfunction in Charcot Marie Tooth 2B Peripheral Sensory Neuropathy. Commun Biol 5, 717 (2022). https://doi.org/10.1038/s42003-022-03632-1
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Artificial Intelligence Methodology in Structural Biology
Publishing Model: Hybrid
Deadline: Nov 30, 2026
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in