Mitochondrial single-cell multi-omics for lineage tracing and mitochondrial genetics
Published in Protocols & Methods
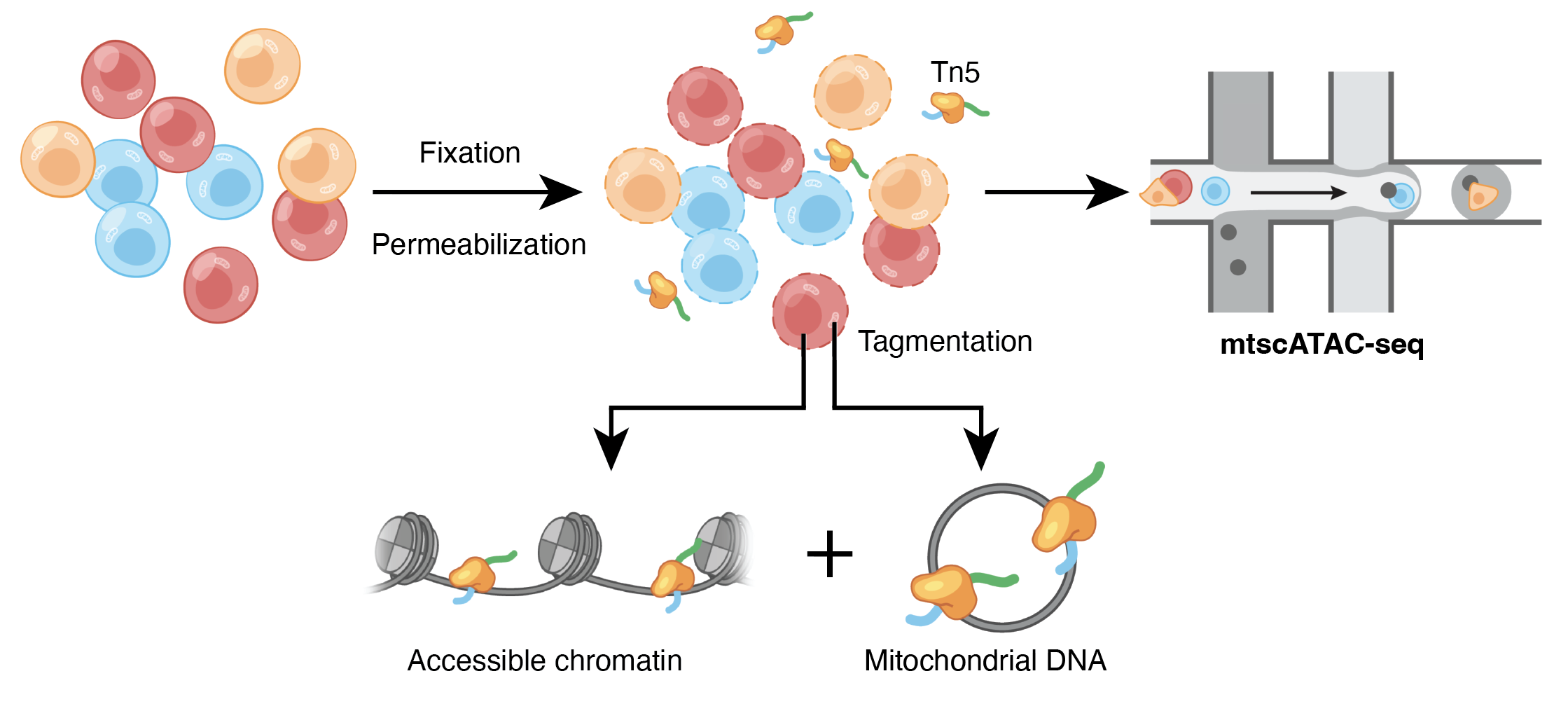

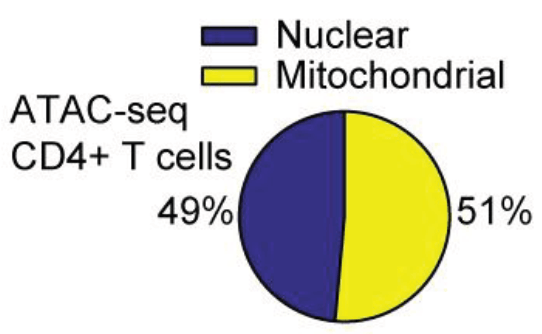
With thousands of citations, the Assay for Transposase Accessible Chromatin by sequencing (ATAC-seq) is one of the most widely-used genomics protocols, even a decade after its first report1. Though the word “mitochondria” does not appear in the original publication, early adopters of this approach know that mitochondrial DNA (mtDNA) could be quite abundant. This is because mtDNA is not densely compacted with histones around nucleosomes, readily enabling the Tn5 transposase to tagment mtDNA. For example, in CD4+ T cells, early ATAC-seq libraries showed >50% of reads mapping to mtDNA2 (Figure 1).
As the primary use case for ATAC-seq is profiling accessible chromatin landscapes in nuclear genomes, many ATAC-seq users sought to discard mtDNA as an unwanted ‘nuisance’ via approaches like CRISPR/Cas93 or revised protocols (e.g., Omni-ATAC)4, to reclaim half of their sequencing reads. We, on the other hand, took a different approach, realizing that mtDNA is somatically hypermutated and could serve as natural genetic markers of cells to resolve clonal and lineage relationships.
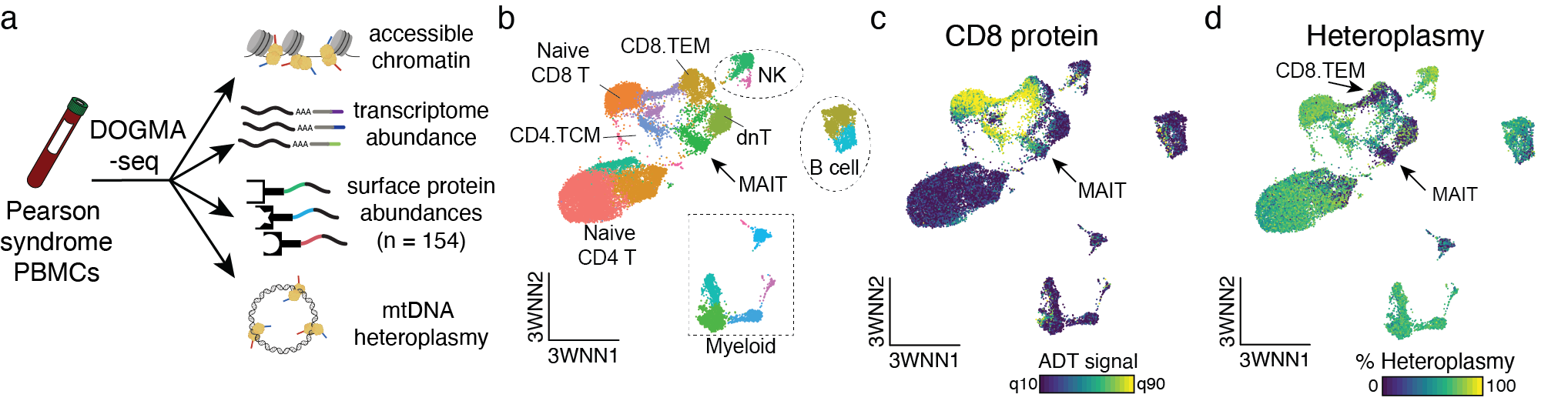
Fast-forward to 2019 and 2020, when we provided the proof-of-concept to leverage single-cell sequencing to detect somatic mtDNA mutations as clonal markers5 and developed mitochondrial single-cell ATAC-seq (mtscATAC-seq), the first readily scalable assay to effectively enable whole mitochondrial genome sequencing and accessible chromatin profiling in ten to hundred thousand single cells6. In the context of hematology and immunology, we routinely apply mtscATAC-seq to decipher clonal population dynamics of human hematopoiesis6 (Figure 2), within the innate immune system7, or study mitochondrial genetics, such as the selection of pathogenic mitochondrial DNA (mtDNA) variants in human T cells8,9 (Figure 3), also using variant workflows of the 10x Genomics Multiome kit (e.g., DOGMA-seq)10.
 Schematic of experimental design. CD34+ HSPCs and peripheral blood mononuclear cells (PBMCs) were profiled from a healthy donor using mtscATAC-seq. (b,c) Two-dimensional embedding using UMAP and colored by cell state annotation. (d) Select cell clone and distribution of clonal members across CD34+ and PBMC chromatin accessibility profiles (as in b,c). (e) Percentage heteroplasmy (log10 scale) of somatic mtDNA variants and respective allele frequencies in pseudobulk CD34+ HSPC (x-axis) and PBMC populations (y-axis), providing evidence of their stable propagation from the CD34+ progenitor to the differentiated PBMC compartment.")
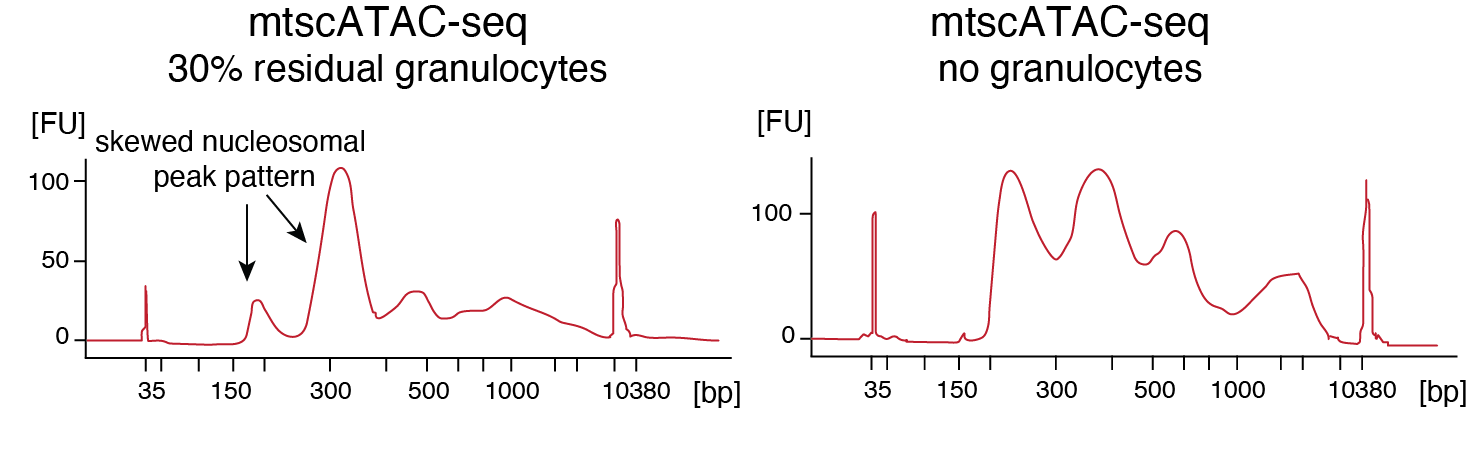
In our Nature Protocols publication, we now compiled our experiences of the past few years to describe the mtscATAC-seq method in detail, including different strategies to enrich your cells of interest via sorting as well as guidance to apply it to solid tissues, which has worked for example very well for us in human ovarian cancer. As neutrophils have Neutrophil Extracellular Traps (NETs) and a peculiar chromatin structure, we highly recommend excluding them via sorting, as they will skew your library distribution and negatively affect your data (Figure 4).
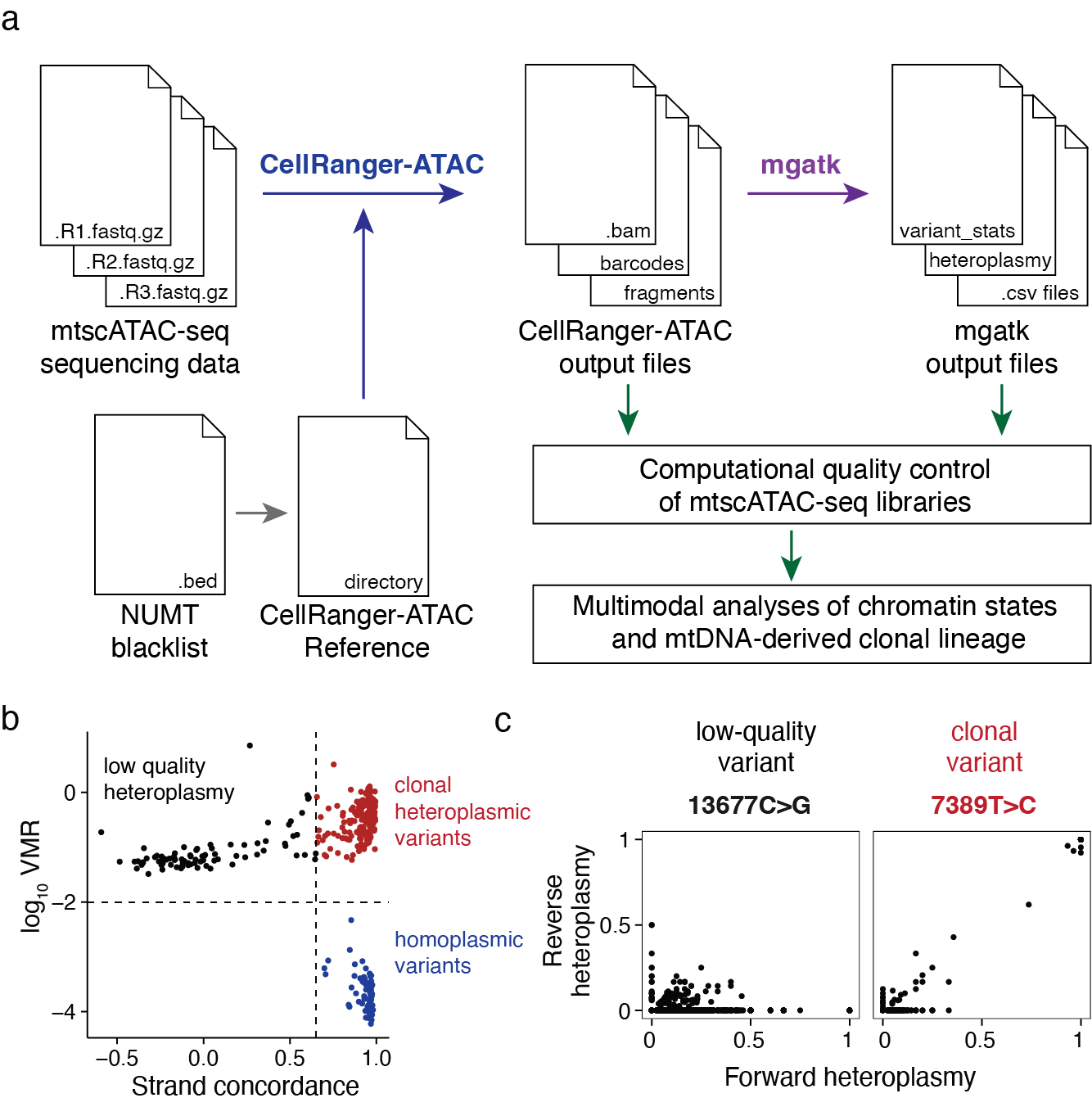
We also dive deeper into the integrated processing with 10x Cellranger-ATAC and our updated computational mgatk workflow for effective variant calling from mtDNA (Figure 5), including generating a custom reference genome, which is essential given the high homology of many regions in the mitochondrial genome with regions in the nuclear genome (NUMTs).
We really hope our protocol helps get you started on mtscATAC-seq or refine your workflow further. There have already been various applications by the community in the human retina11, murine leukemias12, and further adaptations of the concept and methodology in scATAC-seq13 and scRNA-seq space14,15. We would love to hear about your experiences and any feedback and comments are very welcome!
References
1. Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. & Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013)
2. Corces, M. R. et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 48, (2016).
3. Montefiori, L. et al. Reducing mitochondrial reads in ATAC-seq using CRISPR/Cas9. Sci. Rep. 7, (2017).
4. Corces, M. R. et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017).
5. Ludwig, L. S. et al. Lineage Tracing in Humans Enabled by Mitochondrial Mutations and Single-Cell Genomics. Cell 176, (2019).
6. Lareau, C. A. et al. Massively parallel single-cell mitochondrial DNA genotyping and chromatin profiling. Nat. Biotechnol. 39, 451–461 (2020).
7. Rückert, T., Lareau, C. A., Mashreghi, M.-F., Ludwig, L. S. & Romagnani, C. Clonal expansion and epigenetic inheritance of long-lasting NK cell memory. Nat. Immunol. 23, 1551–1563 (2022).
8. Lareau, C. A. et al. Single-cell multi-omics reveals dynamics of purifying selection of pathogenic mitochondrial DNA across human immune cells. bioRxiv 2022.11.20.517242 (2022)
9. Walker, M. A. et al. Purifying selection against pathogenic mitochondrial DNA in human T cells. N. Engl. J. Med. 383, 1556–1563 (2020).
10. Mimitou, E. P. et al. Scalable, multimodal profiling of chromatin accessibility, gene expression and protein levels in single cells. Nat. Biotechnol. 39, 1246–1258 (2021).
11. Mullin, N. K. et al. Non-random distribution of mitochondrial m.3243A>G heteroplasmy in human retina and its impact on cellular phenotype. bioRxiv 2022.06.20.496449 (2022)
12. Penter, L. et al. Mitochondrial DNA Mutations as Natural Barcodes for Lineage Tracing of Murine Tumor Models. Cancer Res. OF1–OF6 (2022).
13. Myers, R. M. et al. Integrated Single-Cell Genotyping and Chromatin Accessibility Charts JAK2V617F Human Hematopoietic Differentiation. bioRxiv 2022.05.11.491515 (2022)
14. Miller, T. E. et al. Mitochondrial variant enrichment from high-throughput single-cell RNA sequencing resolves clonal populations. Nat. Biotechnol. 40, 1030–1034 (2022).
15. Gier, R. A. et al. Clonal cell states link Barrett’s esophagus and esophageal adenocarcinoma. bioRxiv 2023.01.26.525564 (2023)
Follow the Topic
-
Nature Protocols
This journal publishes secondary research articles and covers new techniques and technologies, as well as established methods, used in all fields of the biological, chemical and clinical sciences.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in