Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection
Published in Bioengineering & Biotechnology


In 2013 I got my first independent position, moving from University College London, where I was a Senior Postdoc, to Norwich Medical School, University of East Anglia for an Assistant Professorship. I had spent most of my research career in the area of infectious diseases diagnostics and this was the area I intended to pursue independently. However, all my previous work was based on nucleic acid amplification technologies such as PCR and nucleic acid sequence-based amplification (NASBA). I wanted to drive infectious diseases diagnostics forward and I felt we needed new technology as I could not see PCR replacing microbiological culture. It was around then that Oxford Nanopore Technologies were making a splash in the genomics community and the Professor who hired me, John Wain, highlighted this technology to me. This was the first time I had considered metagenomics as a relevant technology for routine infectious diseases diagnostics as existing technology was too slow and too expensive. So, when ONT put out a call to the community for the MinION Access Programme (MAP), I jumped at the chance. I felt this technology was crucial for my research moving forward, so I got in touch with Zoe McDougall at ONT (through an introduction by Nick Loman) to explain what we were doing and why I felt we would be good early access customers. Zoe was (and still is - along with Leila Luheshi in more recent years) very supportive of our work, and we were lucky enough to get chosen for the first wave of MAP customers and received our first MinION in June 2014. I think it is fair to say that the MinION wasn’t fully optimised on release to the MAP community, but we managed to get some early data that helped us to solve an issue that we had with Salmonella Typhi using the long nanopore reads in combination with Illumina data (Figure 1). We published this study in Nature Biotechnology1 and it was the first MinION application paper published. This paper served to demonstrate the huge potential of the technology and gave my group a profile in the area of nanopore sequencing.
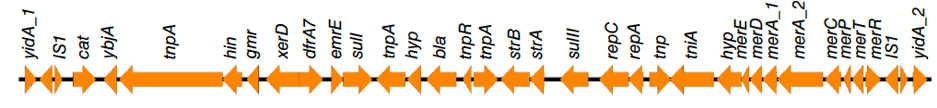
Figure 1: Genetic organisation of a novel S. Typhi chromosomal resistance island resolved using nanopore reads in combination with short read sequence data1.
MinION performance, in terms of sequence accuracy and yield, was improving all the time so we decided it was time to begin utilising it for infection diagnosis (i.e. clinical metagenomics). Back in 2015, large amounts of DNA were required for MinION library preparation (>1µg), which didn’t lend itself well to this application - it was more suitable for bacterial isolate sequencing. However, urine samples from patients with urinary tract infection (UTI) often have high bacterial loads, similar to an overnight culture of bacteria, so we chose UTIs as our first disease for which to apply rapid clinical metagenomics. We had to use urine samples containing >107 CFU/ml and the performance of the early flowcells was highly variable, but we managed to sequence 10 clinical samples and detect the correct pathogens and acquired antimicrobial resistance genes. Key to the success of this application was developing a method to efficiently remove human DNA from the sample. Where there is infection there are leukocytes - each of these leuocytes contains about 1000-fold more DNA than a typical bacterial pathogen e.g. E. coli. The number of leukocytes in urine varies but there can be in the range of 106 per ml, hence there is typically more human DNA in a urine sample than bacterial (typically >10 fold). If only 10% or less of your data is on target, then obtaining the sequencing data you need becomes slow and expensive. Plus, the MInION yields at the time weren’t what they are now, so every read counted (Figure 2).
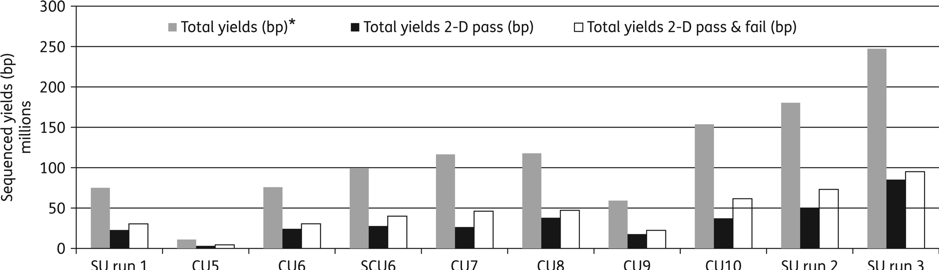
Figure 2: Improvement of MinION sequencing performance and yields (6 hours sequencing run time) over the course of the study. SU, spiked urine; grey bars, total yields (1-D template + 1-D complement) (bp); black bars, total yields 2-D pass (bp); white bars, total yields 2-D pass & fail (bp)2.
The depletion was a combination of differential centrifugation with the Molysis Basic 5 kit (a commercial host depletion method). Neither approach was sufficient alone, but when combined resulted in highly efficient host depletion. The method, however, was relatively slow and expensive. The mature pipeline consisted of host depletion, bacterial DNA extraction, rapid library preparation (15 mins), MinION sequencing and data analysis and took approximately 4 hours from sample-to-result. The UTI study was a useful proof-of-concept to demonstrate the potential of rapid clinical metagenomics and was published in JAC in 2016/172. However, there was still a requirement for high numbers of bacteria, so all potential UTIs could not be tested. Also, the low throughput and high cost of MinION sequencing made it impracticable for implementation for routine UTI diagnosis (requirement to test thousands of samples per week in most clinical microbiology labs).
Two advances were necessary for rapid clinical metagenomics to move forward i.e. rapid, simple, effective, low-cost host depletion and low-input library preparation. In early 2017 Oxford Nanopore released the rapid low input library preparation kit (which required only 1ng (or even pg in our hands) input DNA) and we started developing a rapid saponin based depletion method. We had started a new programme grant on rapid diagnostics for hospital acquired pneumonia in 2016 (the INHALE study - https://www.ucl.ac.uk/inhale-project), and decided that lower respiratory tract infection would be a good application for clinical metagenomics. LRTIs are the leading infectious killer globally and current diagnostics are insensitive and slow. Soon one of the PhD students in my lab, Ms Themoula (Themis) Charalampous, was running our new saponin depletion method on sputum samples provided by the Norfolk and Norwich University Hospital Clinical Microbiology Laboratory and sequencing them with the help of Dan Turner and David Stoddard (the library prep guru) at ONT. This was the beginning of the paper published today in Nature Biotechnology3. We tested the initial pipeline on 40 samples, then optimised and tested on an additional 41 samples. The optimised pipeline has high clinical specificity and sensitivity for the identification of pathogens when compared to culture and PCR and takes approx. 6 hours from sample-to-result (Figure 3). This is approx. 40 hours faster than culture and when implemented will improve how patients with LRTI are managed, reducing morbidity and mortality.
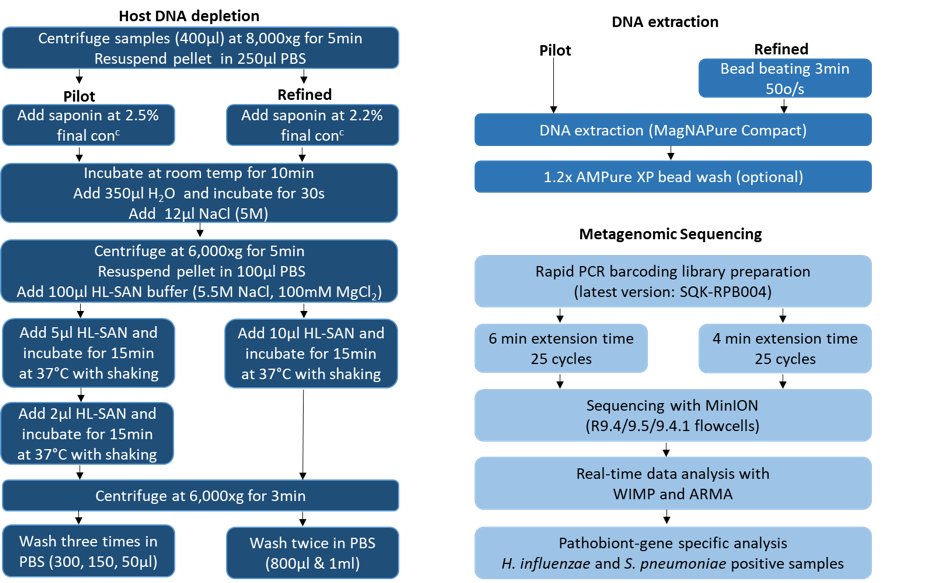
Figure 3: LRI clinical metagenomics pipeline (pilot and optimised methods)3
Challenges remain however, and this is not the perfect test. Accurately predicting antimicrobial susceptibility of pathogens in lung samples that contain commensal flora is challenging and more research needs to be done in this space. Another issue is viral detection. Obviously, viruses are important causes of LRTIs and the current test does not detect them. This is because we pellet the bacteria by centrifugation during the depletion process and the viruses are lost in the discarded supernatant. We have a 2-armed protocol where we keep the supernatant for viral detection but to reach the sensitivity required for respiratory virus detection, whole genome amplification (WGA) is required and this adds time/complexity to the method. Bacteriology and virology are typically separate departments in the clinical microbiology laboratory, with bacteriology focussed on culture and virology focussed on PCR (and other targeted amplification-based methods). I would envisage our method replacing respiratory culture in the bacteriology lab, leaving the virology lab to continue the use of PCR for the diagnosis of respiratory viruses.
Our clinical metagenomics pipelines are constantly evolving and we are in the process of simplifying the depletion process and speeding up the library preparation PCR. We expect to be able to get from sample-to-result in 4 rather than six hours in the near future. As the pipelines mature, we must gather the evidence we need to prove clinical efficacy after which we can begin the process of clinical implementation. We have just finished a diagnostics evaluation trial (INHALE) where we compared our clinical metagenomics method to 2 multiplex PCR tests for the diagnosis of hospital acquired pneumonia. We have recently secured a grant to help implement our laboratory developed test in China in collaboration with a Chinese Diagnostics company (funded by Innovate UK and the Chinese Government). These are the first steps on a long road for just one of our clinical applications. However, I am convinced that clinical metagenomics will become an important diagnostics method in the clinical microbiology in the near future, and may perhaps replace culture as routine practice in the long term.
References
1. Ashton PM, Nair S, Dallman T, Rubino S, Rabsch W, Mwaigwisya S, et al. MinION nanopore sequencing identifies the position and structure of a bacterial antibiotic resistance island. Nat Biotechnol. 2015;33(3):296-300.
2. Schmidt K, Mwaigwisya S, Crossman LC, Doumith M, Munroe D, Pires C, Khan AM, Woodford N, Saunders NJ, Wain J, O'Grady J, Livermore DM. Identification of bacterial pathogens and antimicrobial resistance directly from clinical urines by nanopore-based metagenomic sequencing. J Antimicrob Chemother. 2017;72(1):104-14.
3. Charalampous T, Kay GL, Richardson H, Aydin A, Baldan R, Jeanes C, Rae D, Grundy S, Turner DJ, Wain J, Leggett RM, Livermore DM, O'Grady J. Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection. Nature Biotechnol 2019.
Follow the Topic
-
Nature Biotechnology

A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in