Near-infrared light driven two-photon-absorbing photocatalysis
Published in Chemistry
Near-infrared (NIR) light-driven photocatalysis has a great potential to overcome the limitations of UV/visible light-driven photocatalysis, such as poor penetration through reaction media and competing absorption by species involved in a reaction. Due to the lower energy of NIR light, in order to produce the desired excited state of a photosensitizer, the absorption of two or more NIR photons is mandatory. One strategy to achieve this scenario is triplet-triplet annihilation upconversion. Even though this strategy has been recently employed in organic photocatalysis, its wide application could be limited by its complex nature and the availability of suitable sensitizer/annihilator pairs. An alternative and more straightforward strategy for NIR photocatalysis is through direct two-photon absorption (TPA), wherein a chromophore is capable of simultaneously absorbing two photons in a single step to populate the desired excited state (Figure 1a).
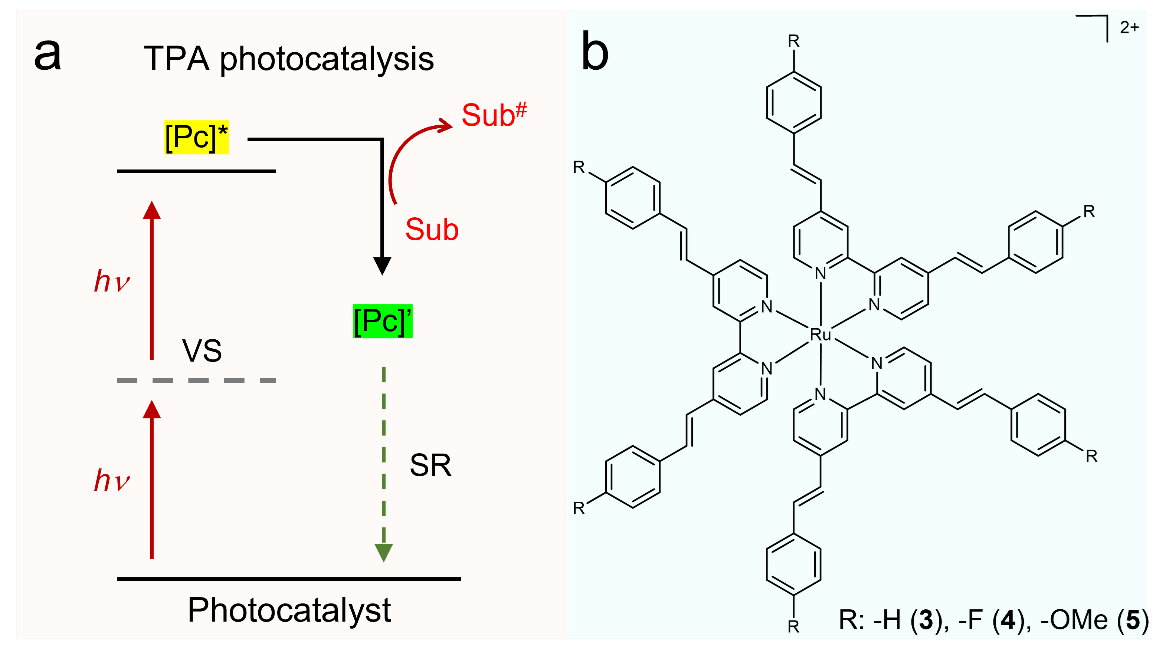
Figure 1. (a) Schematic illustration of two-photon absorption (TPA) photocatalysis. Pc: photocatalyst; SR: sacrificial reagent; Sub: substrate; VS: virtual state. (b) Molecular structures of reported Ru TPA complexes.
Despite the successful utilization of TPA complexes in fields like bioimaging and photodynamic therapy, their application in homogeneous photocatalysis remains very much underexplored, likely due to the small TPA cross sections (σ2) of most photosensitizers suitable for photocatalysis. We envision that a TPA photocatalyst with a large σ2 should be able to drive diverse organic reactions similar to those of one-photon-absorbing photosensitizer, e.g., [Ru(bpy)3]2+ (bpy = 2,2’-bipyridyl), using inexpensive LED as the sole light source. Several studies have demonstrated that octupolar bipyridyl metal complexes coordinated with bisstyryl-substituted bpy ligands exhibit superpolarizability and TPA cross sections two orders of magnitude higher than that of [Ru(bpy)3]2+ in the NIR region.
Herein, we report that a group of Ru complexes ligated by 4,4’-bisstyryl-2,2’-bipyridine (bpyvp) and its close analogues are able to drive a variety of organic transformations upon irradiation at 740 nm following the TPA strategy. A systematic study on the structure-function relationship of these Ru TPA photocatalysts was performed by tuning the ligand ratio between bpy and bpyvp-type ligands and installing electronic-donating (e.g., -OMe) or -withdrawing (e.g., -F) substituents at the para positions of the terminal phenyl groups in bpyvp (Fig. 1b), together with theoretical computation. Comparison of the photophysical and photocatalytic properties of these complexes leads to the following findings. (i) A greater number of bpyvp-ligands in the coordination sphere of these Ru complexes generally lead to better TPA photocatalysis, in agreement with the increased TPA cross section. (ii) Electron-donating substituents at the para positions of the terminal phenyl groups in the bpyvp-type ligands are beneficial for the overall photocatalytic performance, likely due to the improved intra-molecular charge transfer. (iii) These Ru TPA complexes can perform organic photocatalysis in an analogous fashion as those one-photon-absorbing photosensitizers. Both energy-transfer (e.g., 1O2 reactions) and photoredox reactions (e.g., hydrodehalogenation, C-H cyanation, Ni-catalyzed allylation of aldehydes) can be accomplished by these Ru TPA complexes with excellent yields upon irradiation at 740 nm under ambient conditions.
Along the development of novel TPA photosensitizers, it is anticipated that low-energy NIR photons beyond the visible region will be more frequently utilized for organic transformations, complementing the existing UV/visible photocatalysis and also presenting unique advantages, such as deeper penetration into reaction media for large-scale applications and negligible competing absorption by organic substrates.
For further information, please read our published article at https://www.nature.com/articles/s41467-022-29981-3
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in