The paper in Nature Communications is here:
https://www.nature.com/articles/s41467-018-04645-3
Inexpensive and environmentally-benign reactions are two major trends in modern organic synthesis and catalysis. Over the past few decades, platinum-group metals such as ruthenium, rhodium, palladium and iridium have been widely used in organocatalytic reactions, especially coupling reactions and hydrogenations. Recently, first-row transition-metal-catalysts have attracted more attention because of their low cost and abundance. In addition, the development of environmentally-friendly reactions has become increasingly popular.
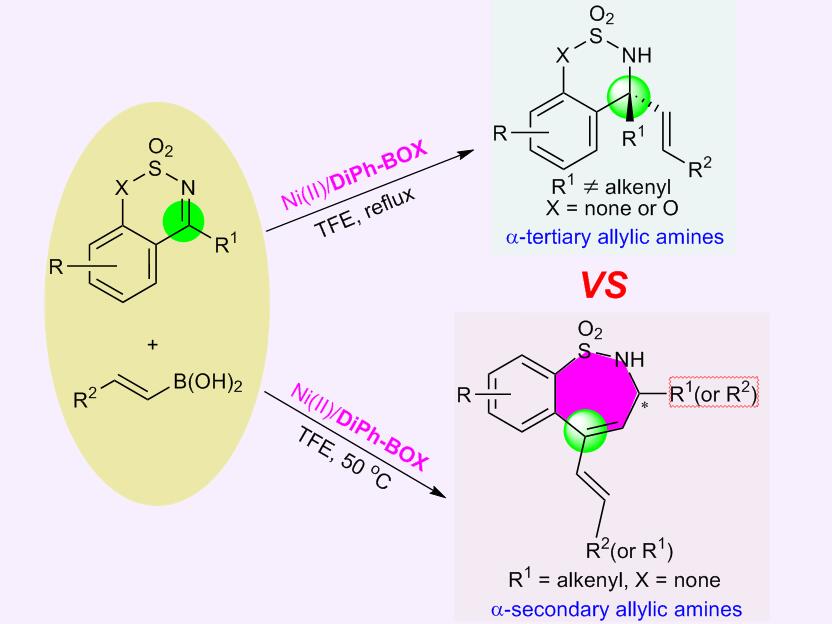
We are interested in the development of transition-metal-catalyzed asymmetric additions of organoboron reagents to imines, the products of which are present in numerous natural products and drugs. Recently, we discovered that first-row transition-metals were able to catalyze these reactions, which are predominantly catalyzed by rhodium or palladium. Furthermore, we discovered that nickel could efficiently catalyze the asymmetric addition of alkenylboronic acids to ketimines. Prior to this, only three ketimine substrates had been successfully reacted with alkenylboronic acid nucleophiles in the presence of a rhodium catalyst.
After thorough screening of the reaction conditions, we found that Ni(II)/DiphBOX was quite effective for this reaction. When the substituents attached to ketimines are alkyl or ester groups, a series of addition products could be obtained. The reactivity and enantioselectivity are both very high and the scope of substrates and alkenylboronic acids is wide. During substrate screening, we desired to test the regioselectivity of this reaction using alkenyl-substituted ketimines. After obtaining the 1H-NMR spectrum of the product, we confirmed that it was most likely the conjugate addition product; however, we had one concern about the spectrum. The peak corresponding to the hydrogen atom of the N-H splits into a doublet, whereas in the conjugate addition product, the hydrogen that is bonded to the nitrogen atom should be a single peak. We initially hypothesized that perhaps the hydrogen on the C=C bond of the additional styrenyl was J4-coupling with the hydrogen of the N-H, but we were not certain. To confirm the exact structure of this product, we cultivated its single crystal and the result was quite surprising - it differed completely from our original hypothesis. The X-ray structure of the product showed that it was a new benzo-seven-membered ring and that a new reaction had been discovered; there are only a few reports concerning the ring-expansion of unstrained cyclic compounds. Next, we systematically studied this reaction. The investigation of the alkenylboronic acid and ketimine scope proved to be very interesting. The experimental results indicated that the site-selectivity of this ring-expansion reaction could be controlled either by the formation of a large π-conjugated system or by steric interactions. The reaction mechanism was also investigated experimentally and computationally with DFT calculations. Mechanistic studies showed that the alkenylation is the enantioselectivity-determining step, while the ring expansion step is a stereospecific process.
Who could have predicted that different substituents could lead to such different results? Chemistry really is full of surprises.





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in