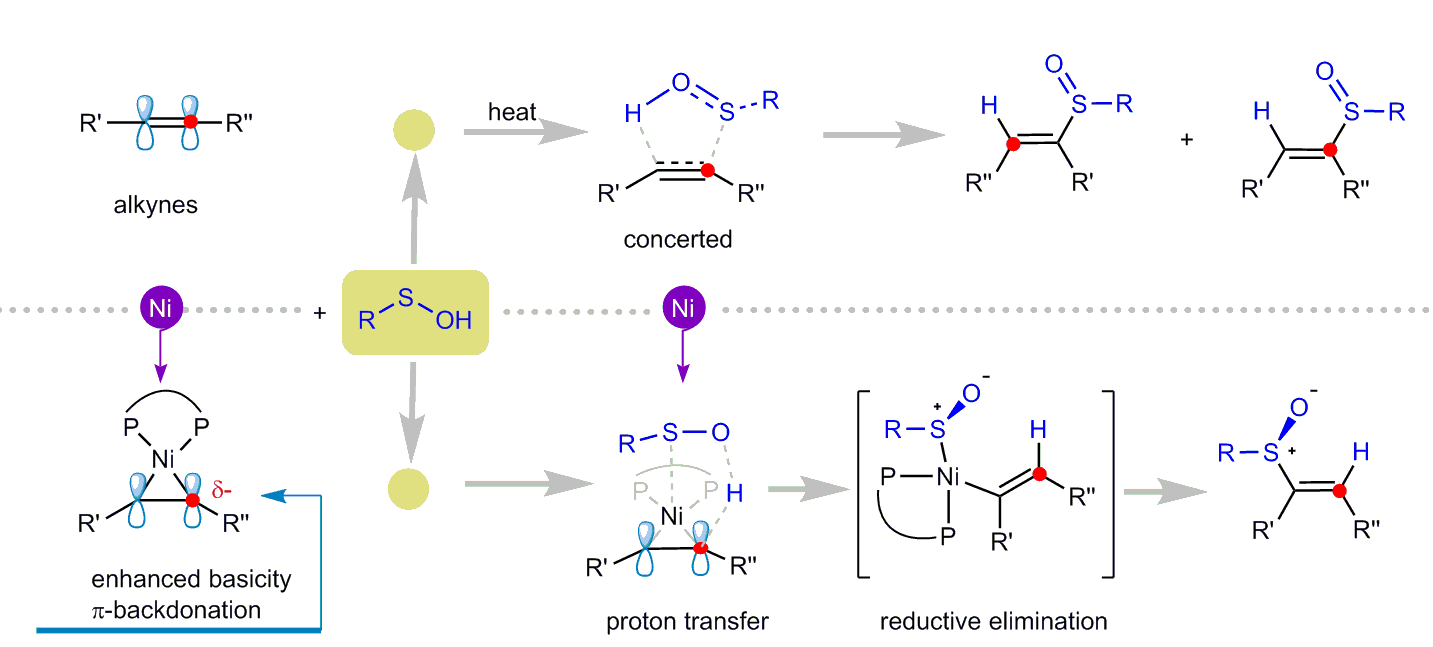

Chiral sulfoxides have found widespread applications in the pharmaceutical industry. Examples of these compounds include Esomeprazole, a proton pump inhibitor, and Modafinil, which is used to treat narcolepsy. Additionally, chiral sulfoxides play a crucial role as ligands in asymmetric synthesis reactions catalyzed by transition metals. The addition reaction to unsaturated bonds is an effective strategy for synthesizing chiral sulfoxides. Although the racemic reaction of in-situ generated sulfenic acid and alkynes was reported as early as 1967 (J. Am. Chem. Soc. 1967, 89, 718.), the corresponding asymmetric catalytic reaction has yet to be achieved. This is mainly due to the high instability of sulfenic acids and their innate reactivity, which are key limiting factors in the development of the reaction. Our research group has been extensively investigating nickel-catalyzed asymmetric synthesis and mechanistic studies. We have proposed a new mechanism model for the asymmetric hydrophosphinylation of conjugated enynes catalyzed by nickel (Chem. Sci. 2022, 13, 4095). We have uncovered a proton transfer mechanism instead of the classic oxidation addition/insertion/reaction elimination process. Based on these findings, we believe that, under appropriate conditions, the asymmetric hydrosulfenation of alkynes to yield chiral vinyl sulfoxide compounds could be achieved by utilizing nickel(0) as the catalyst.
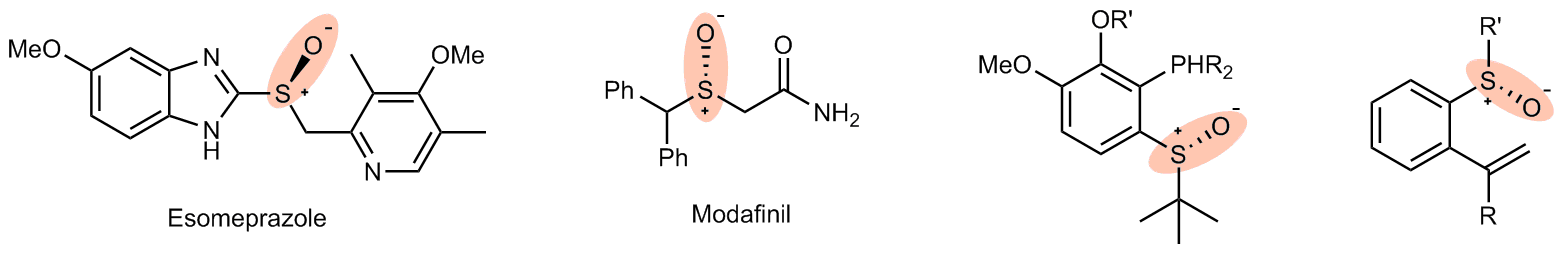
Fig. 1. Representative medicine molecules and ligands.
The nickel-catalyzed asymmetric hydrosulfenation of alkynes poses substantial challenges. Extensive screening of chiral ligands revealed that only strongly electron-donating ones, such as (R,R)-Ph-BPE, can successfully produce the desired product with excellent enantioselectivity. Alkaline additives play a crucial role in controlling the reaction's activity. Weak bases result in significantly slower reaction rates, while the use of strong bases leads to decomposition of the starting materials. Remarkably, the reaction exhibits excellent substrate compatibility. Alkynes with diverse electronic properties, as well as aryl-substituted and alkyl-substituted sulfenic acids, can participate in the reaction. The reaction also proves useful for the post-modification of certain drug molecules and biologically active compounds, affording corresponding products with moderate to high yields and exceptional enantioselectivity. Furthermore, the resulting chiral vinyl sulfoxides can be further transformed into various structurally complex compounds, thus demonstrating the practicality of the strategy.
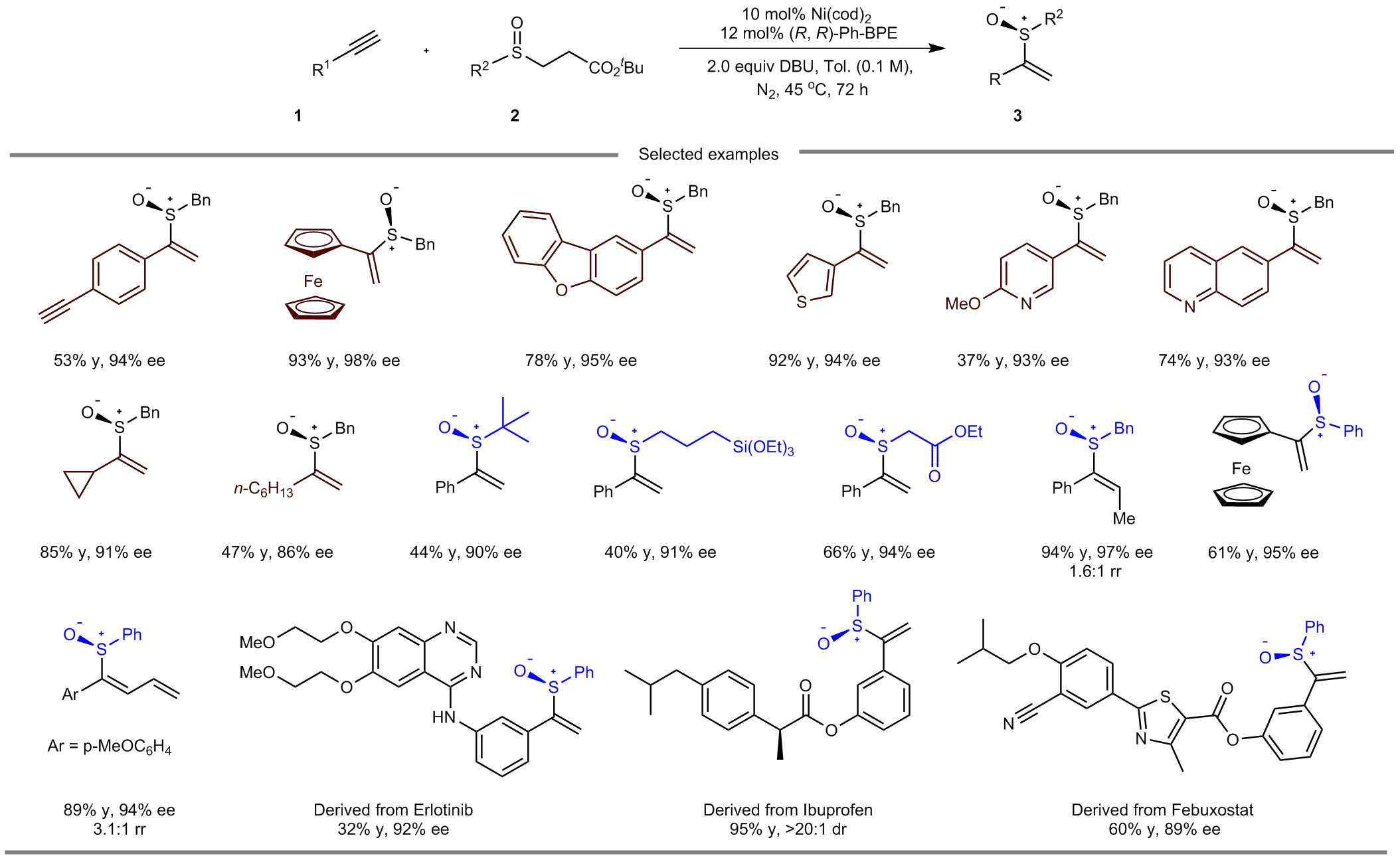
Fig. 2. Selected examples.
Control experiments and density functional theory (DFT) calculations, conducted in collaboration with Prof. Gang Lu of Shandong University, have shed light on the mechanism of the reaction. It has been determined that the reaction proceeds through a nickel-mediated innersphere proton transfer process. Comparison of the Natural Population Analysis (NPA) charges reveals that the coordination of the alkyne with nickel enhances the electronic density of the alkyne through π-backdonation (Bull. Soc. Chim. Fr. 1951, 18, C79; J. Chem. Soc. 1953, 2939.; J. Chem. Soc. 1955, 4456.). This leads to a more negatively charged terminal carbon, indicating a stronger basic site. Furthermore, the coordination of the sulfenic acid with nickel through sulfur further augments the basicity of the alkyne carbon. Concurrently, electron transfer from sulfur to the alkyne renders the proton on the sulfenic acid more acidic compared to the free form. These factors collectively facilitate the subsequent protonation process. The nickel catalyst demonstrates the ability to activate both the alkyne and the coordinating element reagent, thus providing support for the design of other asymmetric hydrofunctionalization reactions involving heteroatoms.
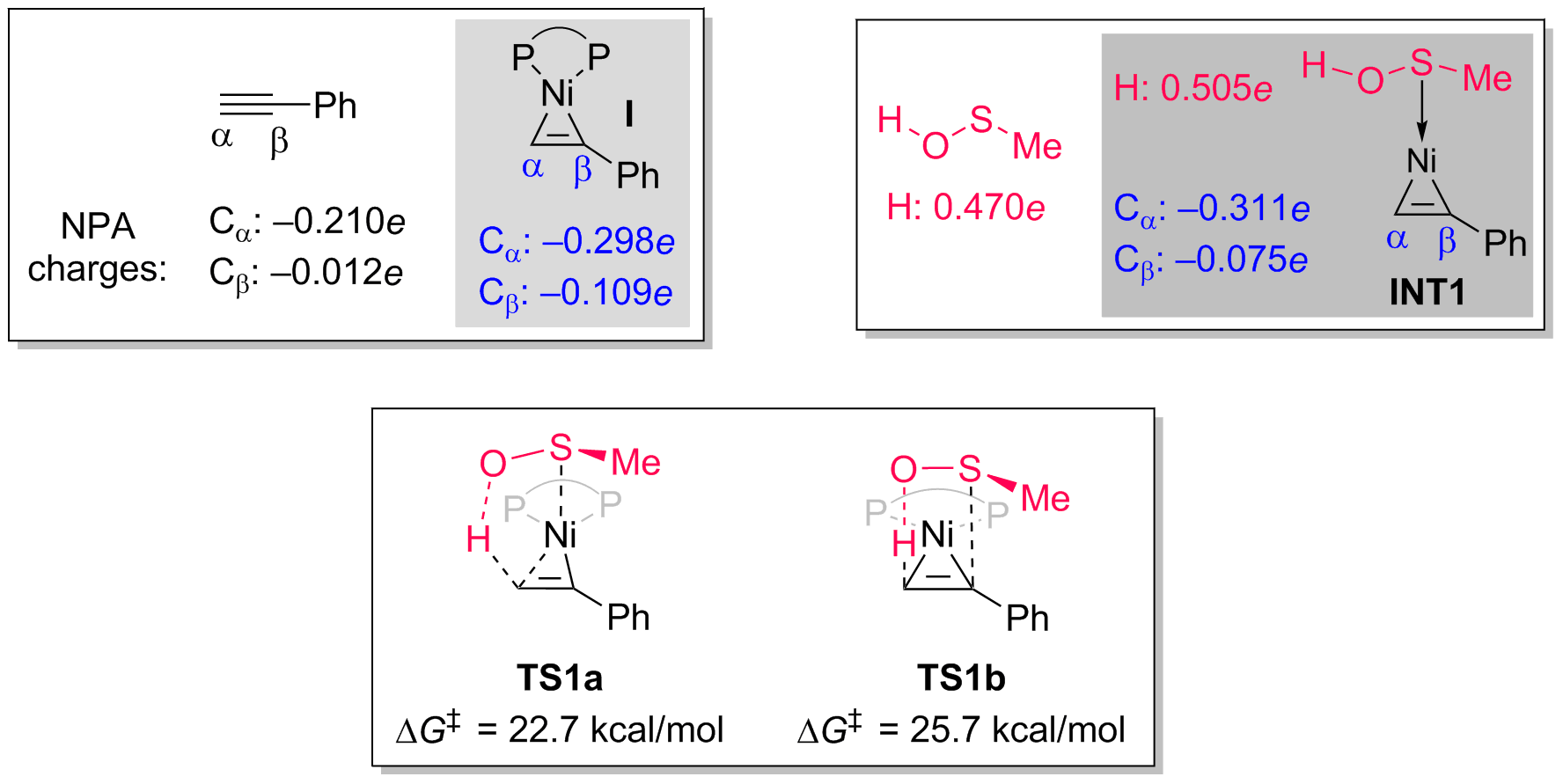
Fig. 3. Computational studies.
More details of this work can be found here: “Nickel-catalysed enantioselective hydrosulfenation of alkynes” in Nature Catalysis. (https://www.nature.com/articles/s41929-023-00966-9)
Follow the Topic
-
Nature Catalysis
This journal brings together researchers from across all chemistry and related fields, publishing work on homogeneous catalysis, heterogeneous catalysis, and biocatalysts, incorporating both fundamental and applied studies.





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in