NTRK activation in cancer is more than fusions
Published in Cancer and Genetics & Genomics
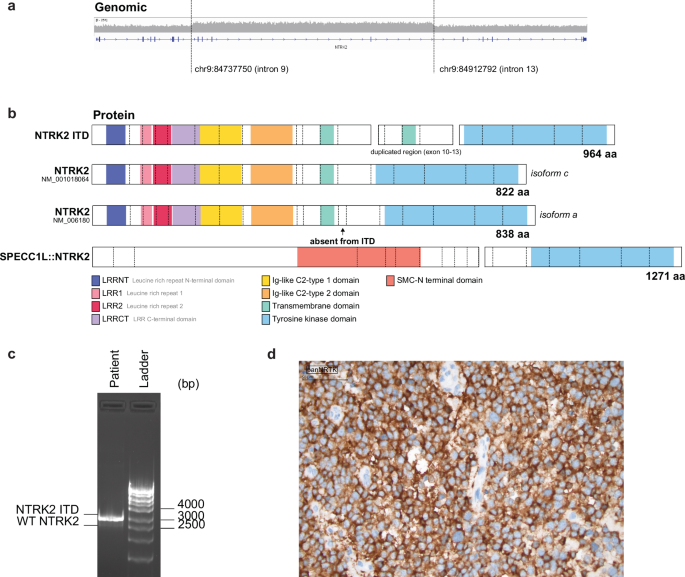
The ZERO Childhood cancer program is Australia's national precision medicine program for paediatric cancer. ZERO has shown that comprehensive genomic analysis and the early use of targeted therapies guided by this analysis, can dramatically alter the survival if children with high-risk cancer. The basis of this improvement is the informed addition of targeted therapies directed against driver lesions within that individual's cancer. Finding NTRK fusions in precision medicine programs like ZERO often causes much excitement, because it means that there are very effective drugs available. But what to do when something new is discovered in the cancer genome, like the NTRK2 internal tandem duplication (NTRK2-ITD) we identified in ZERO? Is this really a driver? Would a cancer with such a lesion respond to a TRK inhibitor? In our report in npj Precision Oncology (linked here), we have shown that NTRK2-ITD is constitutively active, independent of ligand and able to drive cytokine independent proliferation and repress cell death. This is all the more remarkable because this is not a duplication of the Tyrosine Kinase domain, but instead a duplication of the transmembrane domain (Figure 1A and B).

Figure 1. NTRK2 ITD is a TRKB activating event that is sensitive to TRK inhibition
- a) NTRK2 ITD results in an in-frame fusion transcript that encodes a 964 amino acid protein with duplication of exon 10-13, including the transmembrane domain. Created using ProteinPaint. b) NTRK2 ITD promotes IL-3 independent proliferation in Ba/F3 cells, like the SPECC1L::NTRK2 fusion. Plot shows number of viable (determined by trypan blue exclusion) Ba/F3 cells cultured without IL-3 over a 10-day period. Data is presented as mean ± SEM (n=3). c) Schematic of wild-type NTRK2 and the proposed mechanism for NTRK2 ITD activation. We propose that NTRK2 ITD retains the typical structure of WT NTRK2 with the addition of a transmembrane domain (TM, shown in pink) that may span the membrane into the cytoplasmic surface. The addition the presence of additional tyrosine sites (Y534 and Y658) and duplication of the alpha-helix transmembrane domain may mediate ligand-independent dimerization and transphosphorylation of NTRK2 ITD monomers. Created in BioRender. d) Ba/F3 cells transformed by NTRK2 ITD are sensitive to the TRK inhibitor, repotrectinib. Plot shows dose response curves of Ba/F3 cells treated with repotrectinib, where the black dotted line depicts IC50 values. Cells were screened in technical triplicates in three biologically independent experiments and data is presented as mean ± SEM (n=3)
NTRK2-ITD does not function exactly like a fusion, as it is a membrane bound oncoprotein, and it seems likely that a tyrosine residue in the duplicated section, now located intracellularly, results in a conformational change that favours transphosphorylation and activation of NTRK2 (Figure 1C). That possibility is still up for debate. What is not questionable is that NTRK2-ITD signalling can be effectively blocked by existing TRK inhibitor drugs. The implications are important. First, patients with tumours bearing NTRK-ITDs could benefit significantly from the addition of TRK inhibitors to their treatment regimens. However, standard testing would not necessarily detect these lesions, because they are designed to find NTRK fusions. Even if identified, the primary indication for TRK inhibitors - NTRK fusions - means that patients with ITDs might not have the choice to receive these therapies. An important purpose of our work to define the biology of ITDs involving NTRK2, is to help make the case for the inclusion of these lesions in the indications for TRK inhibitors. To be sure, NTRK-ITDs are rare, but they are recurrent. Learning how patients with these drivers respond to targeted therapy is the next step.
Follow the Topic
-
npj Precision Oncology
An international, peer-reviewed journal committed to publishing cutting-edge scientific research in all aspects of precision oncology from basic science to translational applications to clinical medicine.

Related Collections
With Collections, you can get published faster and increase your visibility.
Minimal Residual Disease and Circulating Tumor DNA Dynamics in Personalized Cancer Treatment
Publishing Model: Open Access
Deadline: Mar 12, 2027
Demystifying Rare Breast Cancers
Publishing Model: Open Access
Deadline: Jul 16, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in