One stop shop for the analysis of Capture Hi-C data
Published in Protocols & Methods

Hi-C is a widely-used method for profiling chromosomal interactions. Chromosomal interactions, such as chromatin looping (which brings regulatory elements in the genome close together in 3D space), are particularly important for gene expression control. As such, profiling chromosomal interactions is important for understanding gene regulation in any biological setting.
The Hi-C method uses formaldehyde to “fix” the DNA in its 3D conformation within the nucleus, enabling the DNA regions to be identified using next-generation sequencing. This method works very well for looking at the overall architecture of the genome, but it is not ideal for examining fine-scale interactions. This is because Hi-C samples are very complex, and so a great deal of sequencing would be required to detect individual interactions amongst the rest of the information.
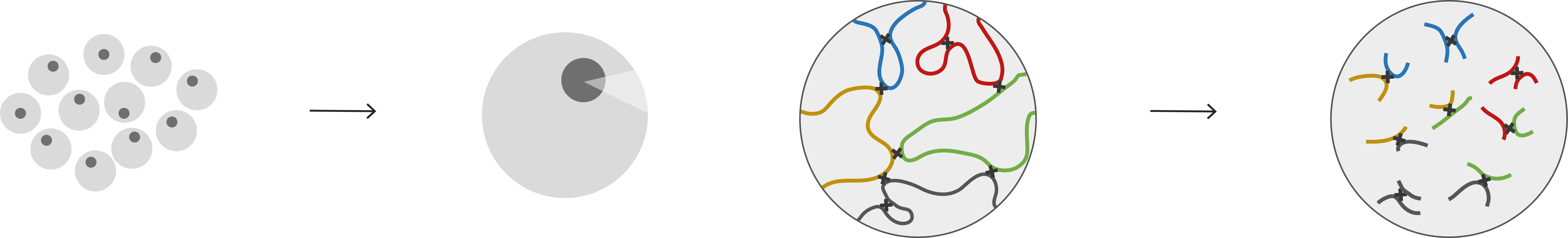
This problem is addressed by Capture Hi-C, which “captures” specific regions of interest within the Hi-C sample (Figure 1). This enrichment step increases our power to detect fine-scale interactions involving those regions - which could be gene promoters, disease-associated loci or any other desired genomic element. Ourselves and others have widely used Capture Hi-C to detect the dynamics of regulatory chromosomal interactions in a variety of different cell types and conditions.
)")
Figure 1. The CHi-C library is generated by performing capture on a Hi-C library followed by paired-end sequencing. (Adapted from Freire-Pritchett et al. 2021)
Capture Hi-C data has some challenging statistical properties that differ from standard Hi-C. For example, the interactions are “asymmetric” because the hundreds to thousands of captured viewpoints are outnumbered by the up to millions of potential interacting other ends. In addition, the regions of interest may have been captured with differing levels of success, which affects the background of the experiment. Therefore, the data has unique statistical properties that require a specialised tool for detecting true signals.
The pre-processing of Capture Hi-C data starts with read alignment and quality control. We recommend using the HiCUP pipeline, developed by Steven Wingett at the Babraham Institute (Wingett et al. 2015). The recent shift in Hi-C to the use of shorter fragments (generated by more frequently cutting restriction enzymes) means that more than one ligation junction may be encompassed within a single read (especially when sequencing long reads). Working together with Steven, we developed a shiny new version of HiCUP (“HiCUP Combinations”), which exploits this scenario. In short, if a read pair contains three interacting genomic loci (X, Y and Z), HiCUP Combinations will detect all potential pairwise interactions between them (X-Y, Y-Z and X-Z). We provide a detailed procedure on how to run both modes of HiCUP in our Nature Protocols article.
The next crucial step in Capture Hi-C data analysis, interaction calling, requires very careful handling. To detect significant chromosomal interactions in a robust manner Jonathan Cairns, Paula Freire-Pritchett and Mikhail Spivakov developed CHiCAGO (Capture Hi-C Analysis of Genomic Organisation), which then became a golden standard for Capture Hi-C data analysis (Figure 2) (Cairns et al. 2016). To do so, CHiCAGO incorporates a bespoke statistical model, background correction and multiple testing procedures.
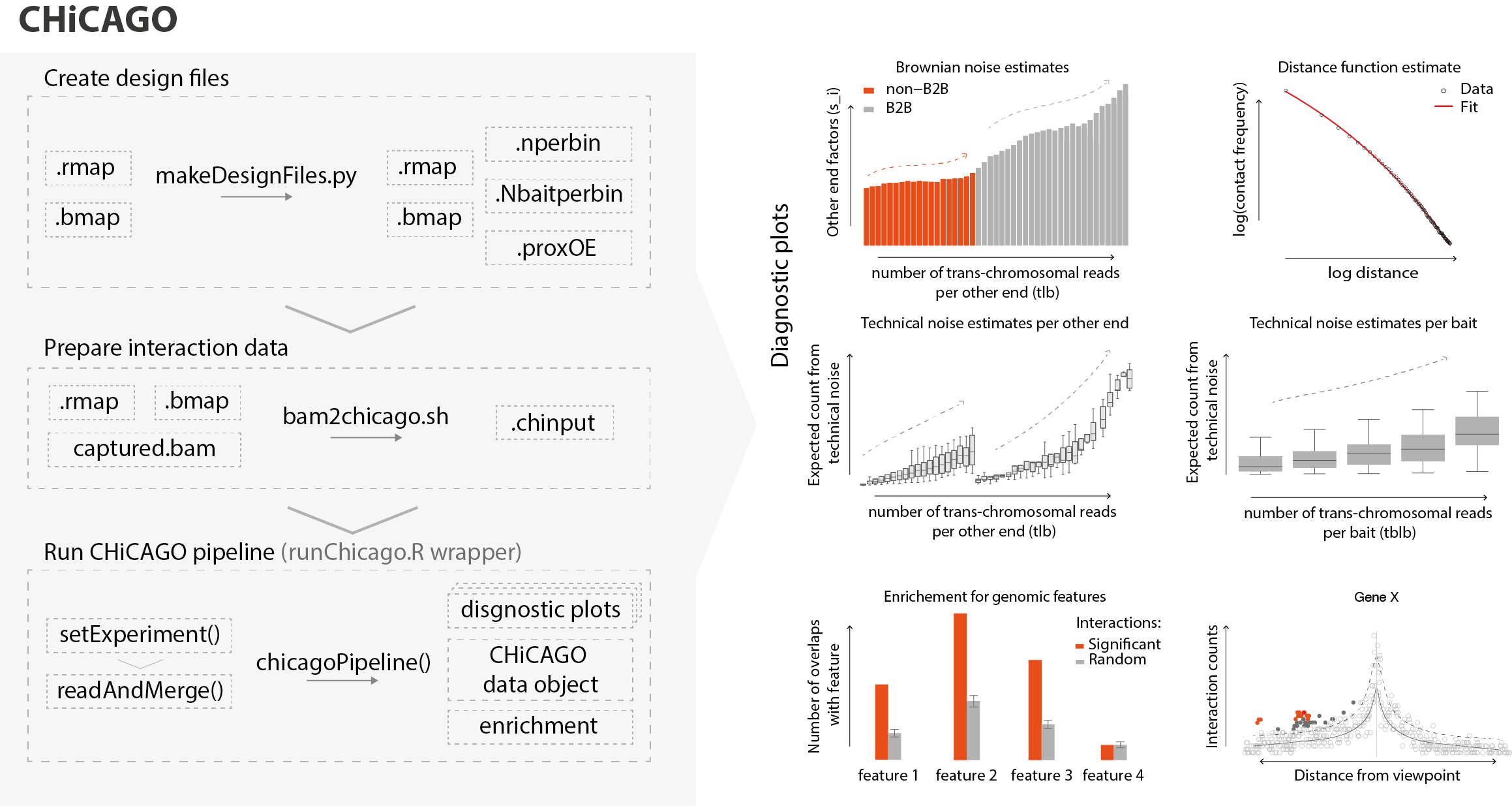
In our detailed protocol, published in Nature Protocols, we provide an extensive guide for users, which we hope will make analysing Capture Hi-C data more accessible for scientists, particularly those who are only just starting with this technique or with bioinformatics data analysis. We share our experience and provide loads of new tips, tricks and scripts to tune the parameter settings of the analysis pipeline to account for data sparsity and experimental design. In particular, we tackle how to analyse data that was generated using more frequently cutting, 4-cutter restriction enzymes, such as DpnII.
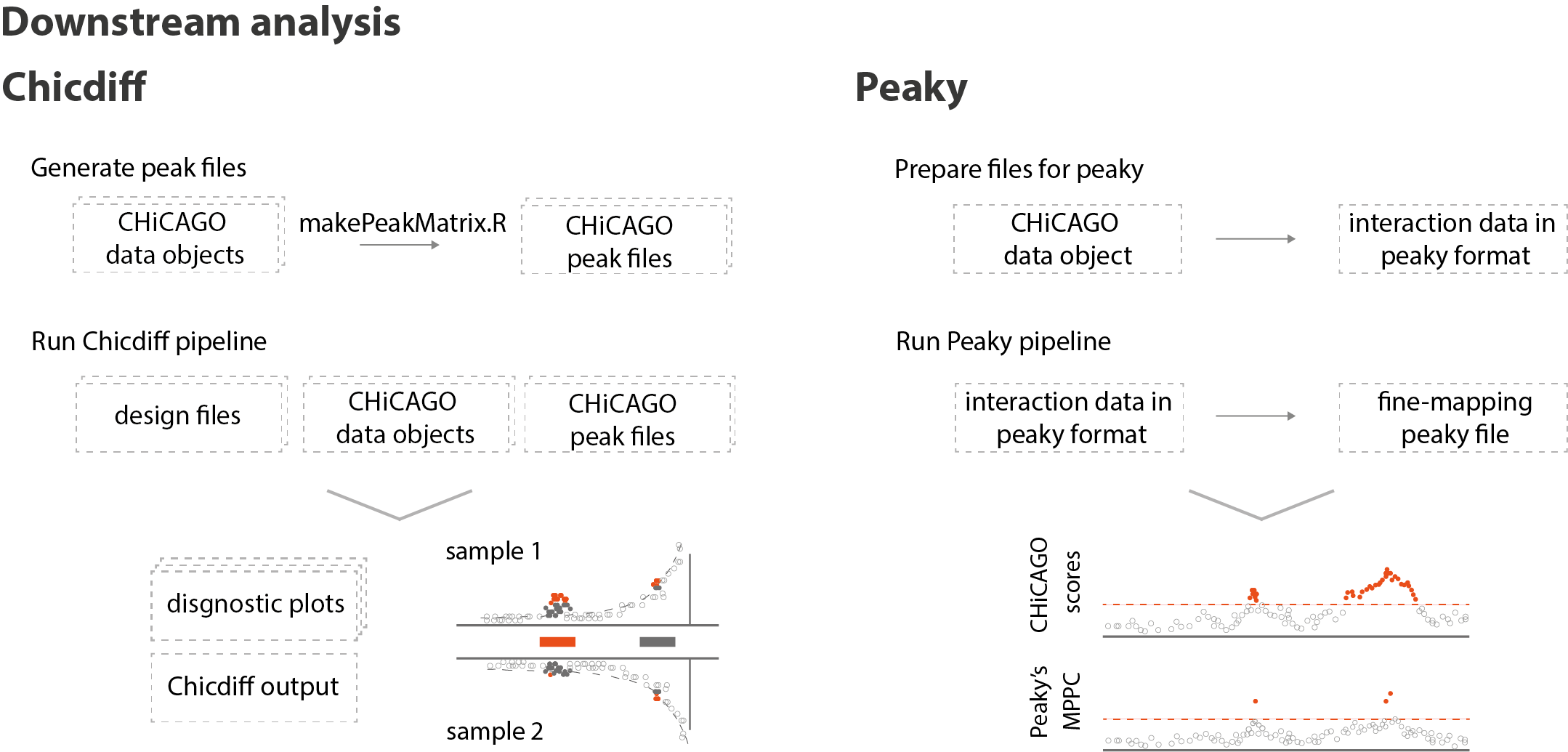
In addition, we describe two companion tools to CHiCAGO that users may wish to use to further explore their results (Figure 3). The first, Chicdiff, is our recently published method for detecting chromosomal interactions that are different between conditions in Capture Hi-C data (Cairns et al. 2019). In our Nature Protocols article, we provide detailed instructions for running this. The second tool, Peaky, was developed in a team of our longstanding collaborator Chris Wallace at the Biostatistics Unit at the University of Cambridge. Peaky is a separate method for fine-mapping chromosomal interactions, which can be used to generate a smaller set of “causal” interacting fragments when the threshold for true interactions is exceeded (Eijsbouts et al. 2019). For our Nature Protocols we worked together with Chris Wallace and Chris Eijsbouts to develop a straightforward way to use their method as part of the CHiCAGO workflow.
Check out our step-by-step guide here: https://www.nature.com/articles/s41596-021-00567-5 with all the code freely available on github and bitbucket.
This work is a result of an amazing collaborative effort and we would like to thank our co-authors for their invaluable contributions!
Happy capturing!
References
Follow the Topic
-
Nature Protocols
This journal publishes secondary research articles and covers new techniques and technologies, as well as established methods, used in all fields of the biological, chemical and clinical sciences.





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in