PheSeq: How Bayesian Deep Learning Conceptualizes the Gene-Disease Associations and Bridges ’em with P-values?
Published in Genetics & Genomics

"This study introduces PheSeq, a Bayesian deep learning model designed to integrate p-value data from sequence analysis with phenotype descriptions from literature and network data. It improves the robustness and interpretability of gene-disease association studies."
Published in Genome Medicine
Apr 16, 2024
Behind the paper [1]: (https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-024-01330-7)
Highlights:
- The Bayesian deep learning framework successfully bridges the phenotype description perception and association significance(p-value) in the gene-disease association studies.
- Deep learning is used to derive embeddings for phenotype description from literature and network data.
- The framework treats the p-value as a weak supervised signal in the uncertainty inference.
- A probability graphical model effectively bridges the aforementioned heterogeneous data modalities by activating a switch when there is consistency between the association significance and the phenotype description.
In the scenario of genotype-phenotype association studies, p-values from various sequence analyses such as GWAS and RNA-seq provide a measure of significance. However, these p-values often come with high uncertainty and lack of interpretability.
The proposed PheSeq model addresses these challenges by combining p-value data with deep learning-derived phenotype embeddings from literature and network data, and bridging two types of heterogeneous association data, thus enhancing the robustness and interpretability of the results.
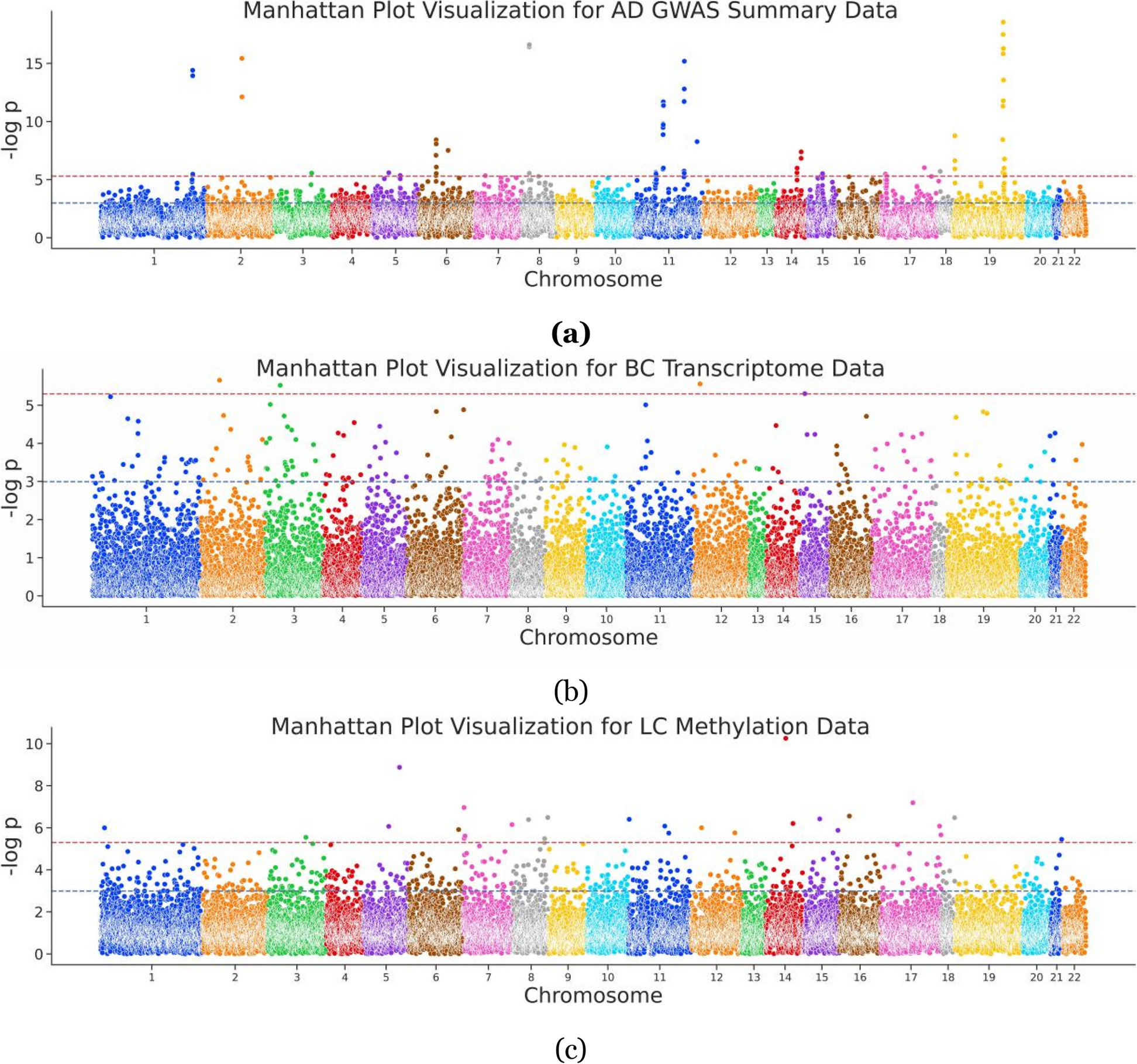
The figure outlines the framework of the Bayesian deep learning model, PheSeq.
|
a. General model input in PheSeq involves p-values for association significance in sequence analysis and phenotypic embeddings for phenotype description from texts or graphs. The associations with p-values are graphically depicted in a Manhattan-style plot. A threshold line with a strict criterion (red line) or a less strict criterion (green line) is then applied. Concurrently, a DL perception module learns the association description of gene-disease association from text or graph. Genes exhibiting significant association descriptions tend to aggregate in the top-left region of the semantic space, as shown in the figure. Analogous patterns emerge in other scenarios. Finally, PheSeq learns the data distributions and performs data fusion for gene-disease associations. b/c Data fusion of association significance and phenotype description for a significant/non-significant gene-disease association by PheSeq. For each gene-disease association, two distinct types of observations, denoted as L for phenotypic embedding and P for p-value, are considered for data fusion. Both sets of observations are input into the PGM inference module, facilitating the learning of dependency relationships among them in conjunction with latent variables. The phenotypic embedding L is initially processed through the DL perception module for semantic training, generating high-quality embeddings denoted as Z. The latent variable T serves a pivotal role in synchronizing the phenotypic embedding data with the p-value data, the latter adhering to a beta distribution indicative of a predisposition toward“small-p-value.” In addition, another latent variable F functions as an association score, establishing connections among model parameters. Conceptually, the switch mechanism activates when both the association significance and phenotype description align, effectively bridging the above heterogeneous data modalities. Part c shows the converse situation, wherein the data indicate non-significance for the gene-disease association. In this case, a uniform distribution is employed to characterize the distribution of the p-value. The remaining configurations of the model remain consistent |
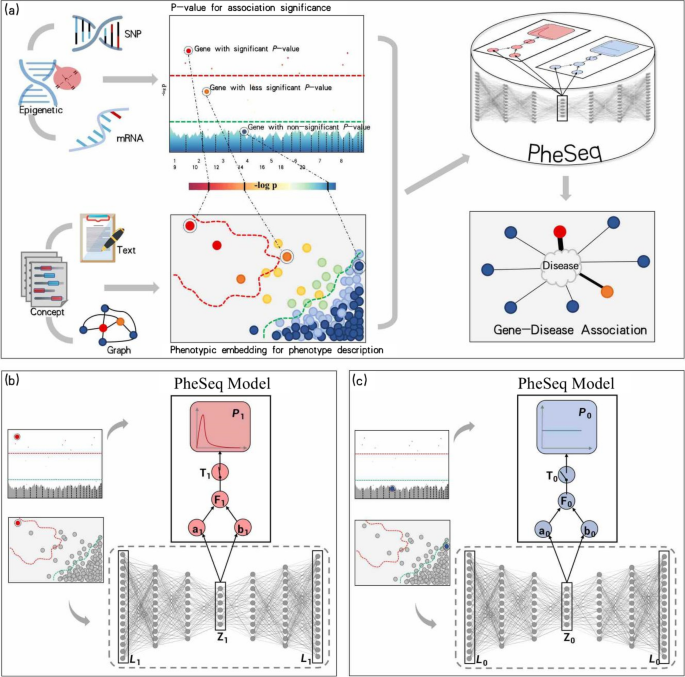
The PheSeq model was tested in three case studies involving Alzheimer’s disease (AD), breast cancer (BC), and lung cancer (LC), using GWAS, transcriptomic, and methylation data respectively. Phenotypic descriptions of the three diseases were collected from disease-related literature downloaded on a PubMed and PMC scale. Sentences that address phenotype description of the gene-disease association are filtered by a biomedical event extraction model on AGAC (Annotation of Genes with Alteration-Centric function changes [2]) corpus.
Finally, PheSeq identified 1024 priority genes for AD and 818 and 566 genes for BC and LC, respectively. Benefiting from data fusion, these findings represent moderate positive rates, high recall rates, and interpretation in gene-disease association studies.
PheSeq holds particular importance in situations where a single sequence analysis may elicit systematic bias and flawed predictions of crucial genes. In such instances, PheSeq serves as an effective tool for establishing a connection between phenotype descriptions and association significance in sequence analysis and helps to recall the significant genes.
In conclusion, this research performs a worth-trying attempt at heterogeneous association data fusion. This framework successfully bridges the phenotype description perception and p-value uncertainty inference. The association significance is utilized as a fine-grained weak signal for the association significance. Overall, it is an inspiring idea to unveil genotype-phenotype associations and investigate the potential relation dependency through data perception, data fusion, and probabilistic inference in a novel Bayesian framework.
Finally, we are delighted to share our work with the scientific community and domain experts in the prestigious journal, Genome Medicine. We sincerely hope that this resource can provide valuable research groundwork and further insights for the community.
References
- Yao, X., Ouyang, S., Lian, Y., Peng, Q., Zhou, X., Huang, F., ... & Xia, J. (2024). PheSeq, a Bayesian deep learning model to enhance and interpret the gene-disease association studies. Genome Medicine, 16(1), 56.
- Wang, Y., Zhou, K., Gachloo, M., & Xia, J. (2019, November). An overview of the active gene annotation corpus and the BioNLP OST 2019 AGAC track tasks. In Proceedings of The 5th workshop on BioNLP open shared tasks(pp. 62-71).
The blog is written by Yanhong He, Fumin Chen, Yawen Liu, Xinzhi Yao, and Jingbo Xia.
- Research Interests
- BioNLP (生物医药自然语言处理)
- Data mining (数据挖掘)
- Bioinformatics (生物信息学)
- Research Projects
- Corpus design and Biomedical knowledge discovery based on BioNLP (语料库设计和基于BioNLP的知识挖掘)
- Data mining for geno-phenotype association (针对表型-基因型关联的生物信息数据挖掘)
Follow the Topic
-
Genome Medicine
This is an open access journal publishing outstanding research in the application of genetics, genomics and multi-omics to understand, diagnose and treat disease, bridging the basic science and clinical research communities.

Related Collections
With Collections, you can get published faster and increase your visibility.
Therapeutic resistance in cancer
Genome Medicine is pleased to invite researchers to contribute original studies focused on the mechanisms of therapeutic resistance in cancer. The Collection will explore topics including drug and immunotherapy resistance, the role of the tumor microenvironment, and innovative strategies to overcome resistance. By advancing our understanding in this critical area, we hope to foster the development of more effective cancer therapies that can improve patient outcomes.
Therapeutic resistance in cancer is a barrier to successful treatment outcomes and patient survival. It encompasses a wide range of mechanisms by which tumors evade the effects of therapies, including chemotherapy, targeted agents, and immunotherapies. Understanding the molecular and cellular basis of resistance is critical for the development of effective strategies to overcome this challenge.
Advances in genomics, proteomics, spatial profiling, and mathematical modelling have enabled researchers to identify key drivers of resistance, paving the way for the development of combination therapies and precision medicine approaches tailored to individual tumors. Exploration of the tumor microenvironment's role in resistance has opened new avenues for intervention, fostering the design of next-generation immunotherapies and targeted agents. This ongoing research is crucial for translating scientific discoveries into tangible benefits for patients facing resistant cancers.
Enhanced understanding of resistance mechanisms could lead to the development of novel combination therapies that preemptively address resistance or exploit emerging transient vulnerabilities, significantly improving treatment outcomes. Furthermore, the integration of advanced technologies such as single-cell sequencing and artificial intelligence in cancer research may facilitate the discovery of new therapeutic strategies, thereby revolutionizing cancer care and management.
In this special issue, guest edited by Dr Luca Magnani and Dr Gao Zhang, we aim to highlight advances in drug and immunotherapy resistance, the role of the tumor microenvironment, and innovative strategies to map resistance development and eventually overcome it. We are inviting the submission of Research, Method, Software, and Guideline manuscripts with significant translational or clinical impact, in all areas of human disease, including:
· Mechanisms of drug resistance in cancer
· Immunotherapy resistance and tumor microenvironment
· Genetic factors in therapy resistance
· Combination therapies to overcome resistance
· Predictive biomarkers for resistance
All manuscripts submitted to this journal, including those submitted to collections and special issues, are assessed in line with our editorial policies and the journal’s peer review process. Reviewers and editors are required to declare competing interests and can be excluded from the peer review process if a competing interest exists.
This Collection supports and amplifies research related to SDG 3: Good Health and Well-being.
This is a sister Collection with Cellular mechanisms and evolutionary processes of drug resistance in BMC Biology; if your research has a focus on biological insights, we encourage you to consider submitting to that Collection.
Publishing Model: Open Access
Deadline: Oct 13, 2026
Precision medicine
Genome Medicine is calling for submissions to a Collection on precision medicine, guest edited by Dr Razelle Kurzrock and Dr Digna R Velez Edwards. Precision medicine represents a transformative approach to healthcare, focused on tailoring medical treatments and interventions to the individual characteristics of each patient. By integrating various data types including genetic, genomic, multi-omic, and environmental, such as patient lifestyle and health, we can better understand disease mechanisms, informing targeted treatment and improving health outcomes.
Advances in multi-omic and sequencing technologies have significantly enhanced our ability to identify biomarkers and stratify patients, thereby facilitating more effective personalized treatment options. As we continue to harness the power of big data and bioinformatics, the application of precision medicine is poised to become more efficient within healthcare systems. Moreover, the evolving landscape of artificial intelligence may enable more accurate predictive models, empowering healthcare professionals to make informed decisions improving patient care.
We are now inviting the submission of Research, Method, Software, Database, and Guideline manuscripts presenting innovative work in the field of Precision Medicine. The collection aims to span a wide variety of disease indications with topics of interest including but not limited to:
• Application of new sequencing technologies
• Multi-omics profiling and approaches
• Integrative analysis and incorporation of healthcare data
• Association studies for risk assessment
• Genomic approaches in clinical practice
• Gene therapy and CRISPR approaches
• Preclinical models for precision targeting
• Biomarker-based monitoring and combination treatments in cancer
• Antibody-drug conjugates and immunotherapy in cancer
• Biomarkers and drug repurposing
• AI and Big data in the clinic
All manuscripts submitted to this journal, including those submitted to collections and special issues, are assessed in line with our editorial policies and the journal’s peer review process. Reviewers and editors are required to declare competing interests and can be excluded from the peer review process if a competing interest exists.
This Collection supports and amplifies research related to SDG 3: Good Health and Well-being.
Publishing Model: Open Access
Deadline: Sep 18, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in