

“While the holy grail remains […] finding a method to catalytically activate and functionalize P4 to yield new organophosphorus compounds, fundamental research lies in developing procedures to control the reactivity of elemental phosphorus.”1
That sentence – written just a few years ago by Ragogna and coworkers – succinctly describes both our motivation, and the biggest problem we faced while pursuing this project. We were seeking to find a direct and catalytic method to functionalize white phosphorus (P4). Why? P4 is the common phosphorus source for almost all organophosphorus compounds, including all manner of industrially-important phosphines and phosphonium salts, among many other species. State-of-the-art processes to functionalize P4 rely on an indirect two-step procedure involving oxidation with toxic chlorine gas to form corrosive phosphorus chlorides. Nucleophilic substitution of the chloride ions then leads to formation of the desired organophosphorus compounds. Far better would be a catalytic and chlorine free method to achieve the same overall transformation in a single step.
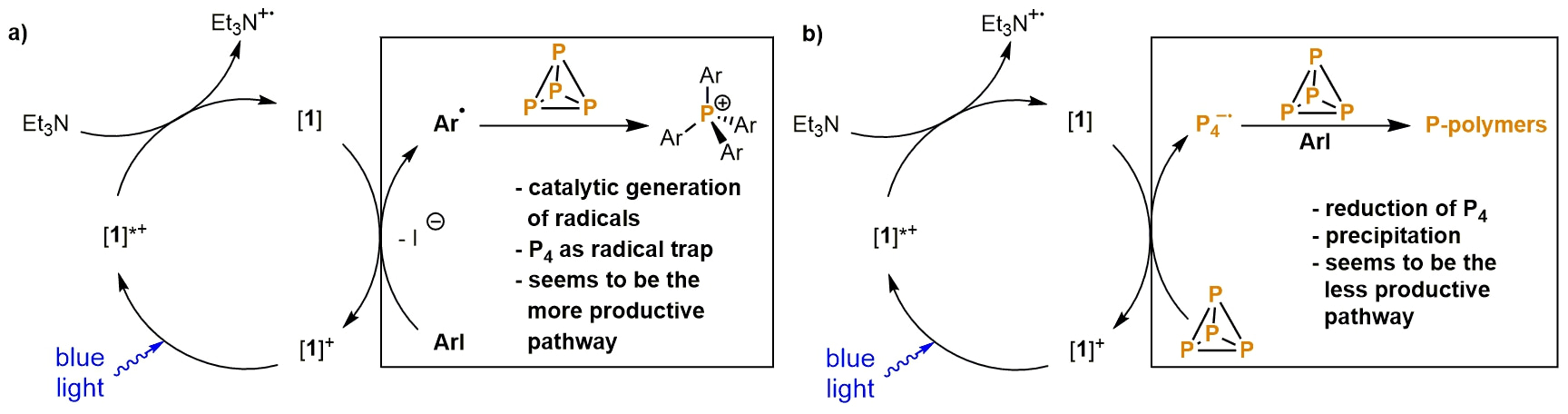
Our initial concept was based on the ability of P4 to serve as an excellent radical trap.2–4 If we could catalytically generate carbon-centered radicals in the presence of P4, this would lead to the formation of new P—C bonds. We chose visible-light-photocatalysis as our “radical supplier”, which was for two reasons. First, P4 is transparent in the visible region, meaning visible light irradiation does not decompose P4. Second, photocatalysis is an unusually mild method to generate radicals, which should minimize any side-reactions with P4 (the chemistry of P4 can be notoriously unselective). We believed that photocatalytic reductive dehalogenation would fit well with our idea (Fig 1a): in the presence of a photocatalyst and a sacrificial electron donor, aryl halides would be reduced to form carbon centred radicals, which could be trapped by P4 to yield arylphosphines or phosphonium salts.
With this idea in mind we began the project, and tried to employ several well-established and well-known photocatalytic systems. Unfortunately, while P4 was consumed in almost all cases, controlling its reactivity proved challenging. In fact, most 31P{1H} NMR spectra and GC-MS analyses did not show any phosphorus containing compounds at all! Instead, a precipitate was almost always formed, and we realised that P4 was probably being reduced directly by the photocatalyst to form the anion P4∙, which then induces polymerization of further P4 (see Fig.1b). Nevertheless, we persisted in our investigation, and were ultimately rewarded with the discovery that using [Ir(dtbby)(ppy)2]PF6 ([1]PF6) as catalyst and Et3N as electron donor solved our problem. The in-situ formed reduced photocatalyst [1] preferentially dehalogenates iodobenzene derivatives to form the desired carbon-centered radicals, and only traces of P4 seem to be reduced. Thus, irradiation of a solution containing [1]+, triethylamine, iodobenzene and P4 in a benzene/acetonitrile mixture with blue LEDs yielded the corresponding tetraarylphosphonium salt, [Ph4P]I. Moreover, substrate screening showed that differently-substituted triarylphosphines and tetraarylphosphonium salts could be synthesized by this method. With these results we were able to validate our reaction design principle, and to demonstrate the first example of an effective catalytic system for the direct organofunctionalisation of P4.
For more information, please read our paper: https://www.nature.com/articles/s41929-019-0378-4
1. Graham, C. M. E., Macdonald, C. L. B., Power, P. P., Brown, Z. D. & Ragogna, P. J. Transition Metal Functionalization of P4 Using a Diarylgermylene Anchor. Inorg. Chem. 56, 9111–9119 (2017).
2. Ghosh, S. K., Cummins, C. C. & Gladysz, J. A. A direct route from white phosphorus and fluorous alkyl and aryl iodides to the corresponding trialkyl- and triarylphosphines. Org. Chem. Front. 5, 3421–3429 (2018).
3. Cossairt, B. M. & Cummins, C. C. Radical synthesis of trialkyl, triaryl, trisilyl and tristannyl phosphines from P4. New J. Chem. 34, 1533–1536 (2010).
4. Barton, D. H. R. & Vonder Embse, R. A. The invention of radical reactions. Part 39. The reaction of white phosphorus with carbon- centered radicals. An improved procedure for the synthesis of phosphonic acids and further mechanistic insights. Tetrahedron 54, 12475–12496 (1998).
Follow the Topic
-
Nature Catalysis
This journal brings together researchers from across all chemistry and related fields, publishing work on homogeneous catalysis, heterogeneous catalysis, and biocatalysts, incorporating both fundamental and applied studies.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in