Predicting the diverse metabolic activities of the microbiome
Published in Microbiology
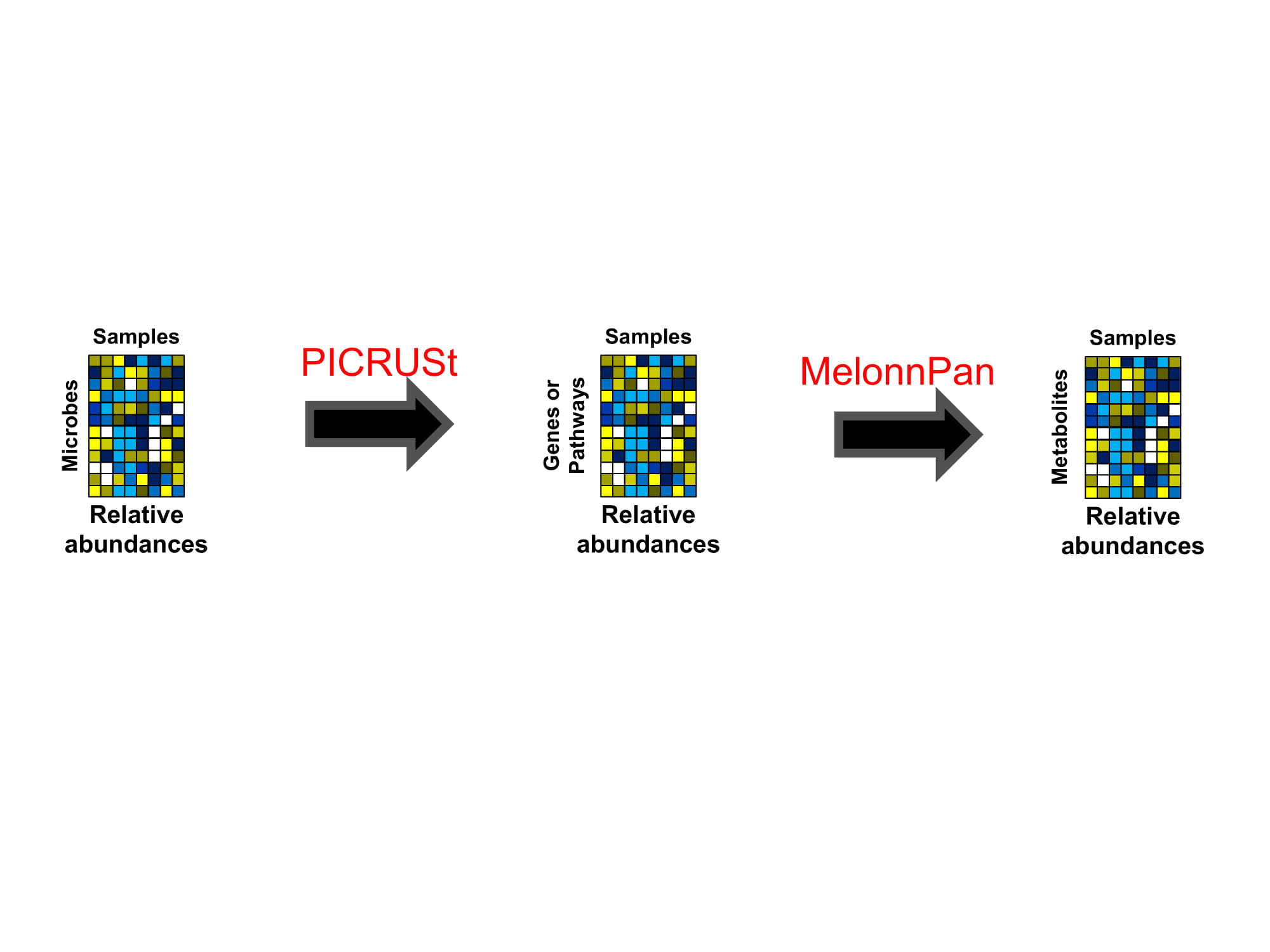

I was a Ph.D. candidate of Biostatistics at the University of Alabama at Birmingham when I read the first article on a tool called PICRUSt - a new computational method that predicts the functional capacity of a microbial community's metagenome from single marker genes (e.g. the 16S ribosomal RNA gene). I was energized by the finding of the paper, i.e. the ability to infer probable functions across many samples from relatively inexpensive 16S data, but was even more excited by the greater consequence of extending this idea beyond gene functions to small molecules - the molecular basis of host-microbiome chemical interactions.
Specifically, inferring the capacity of a microbial community to produce molecules seemed like a promising next step toward the goal of understanding how and why microbiome metabolism affects human health. Microbiome multi-omics was still in its infancy back in 2013, however, and among other obstacles, it was difficult to obtain samples in a format compatible with multiple assays (e.g. metabolomics and metagenomics), as well as costly to generate the associated data. From a computational standpoint, easily identifying these associations purely based on enzymatic roles was also greatly limited by the yet unsaturated reference databases of gene-metabolite-reaction relations.
Things started moving in 2014 when the second phase of the NIH Human Microbiome Project, the Integrative Human Microbiome Project (iHMP or HMP2) was announced. Previous studies had shown strong relationships between microbial sequence features and metabolic abundances, but these studies were limited in scale and scope. HMP2’s population-scale multi-omics profiles offered us a means to re-formulate this problem in a systems biology framework by taking advantage of these potential biological relationships in the freshly generated data from the iHMP cohort.
To this end, leveraging a cross-sectional predecessor of the HMP2 cohort (i.e. PRISM) comprising more than 150 IBD patients and non-IBD controls, we set out to develop a machine-learner that predicts the metabolomic activities or pools of microbial metabolites from metagenomes, based on taxonomic and/or functional profiles. We envisioned this metabolite prediction method as a data-driven approach, rather than relying on known biochemical mechanisms that are still very sparse in microbial communities. Moving forward, this method could also be used to generate new hypotheses and motivate population-scale discovery of novel gene-metabolite associations in metagenomic data collections. In this way, the method would serve as a complementary adjunct to experimental validation studies (similar to the role of predictive functional profiling from amplicon data using PICRUSt).

The method we proposed (MelonnPan: Model-based Genomically Informed High-dimensional Predictor of Microbial Community Metabolic Profiles) is rather straightforward. It first identifies which taxonomic or functional features are predictive of metabolite pools. In new metagenomes, it combines these features to estimate composite, potentially unobserved, metabolomes (Fig. 1). In this prediction process, MelonnPan relies on a model trained on paired metagenomes and metabolites. Specifically, it uses sparse regression to single out a small set of relevant features by progressively setting the contributions of less relevant features to zero. This, in turn, facilitates an interpretable model while also reducing the effect of overfitting in high-dimensional data (i.e. modeling error which occurs when a prediction rule is too closely fit to a limited set of data points). The singled-out features for a particular metabolite may be associated with its abundance by direct enzymatic action, or through other, less-direct mechanisms (MelonnPan is agnostic to such distinctions). Using this ‘predictive metabolomic’ approach, we observed that more than 50% of metabolites were well-predicted in the human gut, which also remained sufficiently accurate in other non-human-gut environments such as coral-associated, murine gut, and human vaginal microbiomes.

Unexpectedly, our human gut model also showed that uncharacterized gene families are the most predictive features in our algorithm, which likely include a combination of genetic markers for specific microbial strains, as well as as-yet-uncharacterized metabolic enzyme classes with potential roles in community metabolism. This is consistent with the proposed role of numerous uncharacterized microbial genes in explaining the vast majority of microbial diversity and function within the human gut. In addition to better understand which microbial genes might process known metabolites, our use of untargeted metabolomic data also allowed us to associate known microbial genes with unidentified metabolites, leading to both biochemical and functional genomic hypotheses for future validation. Finally, we demonstrated that ‘predictive metabolomic’ profiling can recapitulate global metabolomic structure in the IBD metabolome, spanning a broad range of metabolic categories including amino acids, bile acids, fatty acids, and sphingolipids, among others (Fig. 2).
Our study thus provides a cost-effective methodology for integrating microbial gene and metabolite information while suggesting important implications for future microbiome multi-omics studies. While the association of mostly unannotated gene families with metabolite abundances provides promising targets for downstream characterization of the genes themselves, it also calls for large-scale, in-depth validation studies that would enable linking specific microbial chemistry to new individual organisms, genes, and enzymes from meta-omics sources. This ultimately requires a collective effort of integrating computational function prediction with experimental microbial physiological and biochemical mechanistic studies to translate population-level discoveries into new microbiome-based therapeutics and diagnostics.

I am excited to see the methodological implications as well. Improvements in the prediction model might include (i) using a combination of metatranscriptomic and metagenomic predictors while (ii) incorporating specific biological prior relationships (such as phylogeny) into the algorithm. We could also extend the method to (iii) dynamically predict compounds based on longitudinal microbiome profiles (iv) while also addressing the nuances of microbiome data with statistical rigor. I am excited to see these open questions answered in the near future, which is one part of what the field needs to start moving from the bench to the bedside!
The corresponding published paper in Nature Communications is here: www.nature.com/articles/s41467-019-10927-1
Acknowledgments
Post written by the study authors, with special thanks to Eric A. Franzosa, Hera Vlamakis, and the members of the Huttenhower lab.
Himel is a tenure-track Principal Investigator researching at Cornell University's Department of Population Health Sciences. He develops computational methods to generate and validate testable hypotheses that accelerate data-driven discovery. Prior to Cornell, Himel was an Associate Principal Scientist at Merck Research Laboratories and a postdoctoral fellow of Computational Biology and Bioinformatics at Harvard University.
Himel holds a Bachelor of Science (B.Sc.) degree in Statistics from the University of Calcutta and a Master of Science (M.Sc.) degree in Statistics from the Indian Institute of Technology (IIT) Kanpur. He earned his Ph.D. in Biostatistics from the University of Alabama at Birmingham (UAB).
He is a Fellow of the American Statistical Association (FASA) and a recipient of the IISA Early Career Award in Statistics and Data Sciences.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in