ProRefiner: A deep learning model for effective and efficient protein sequence design
Published in Computational Sciences
Structure-based protein design, i.e. Inverse Protein Folding (IPF), aims to design protein sequences from given structures. It has important applications in many protein design and engineering tasks, as adopting a certain conformation is often the prerequisite of performing certain functions. As the name indicates, IPF tackles the reverse process of protein folding, where complex interactions among different components guide the linear protein chain to fold into three-dimensional structure. Therefore, to translate a given structure back into sequences, it is important and helpful to analyze the interplay between residues. Fortunately, deep learning offers an effective approach to model and learn these intricate non-linear many-body relationships. In light of this, we aim to address the question: How can we design a model that can learn and extract residue relationships within 3D structures? Additionally, how can we design a sequence generation pipeline to facilitate the model's representation of residue relationships?
To address these challenges, we propose ProRefiner, a model that can learn to represent global residue interplays effectively and efficiently, and a sequence design pipeline that extracts high quality residue environment to aid the model prediction.

Sequence design pipeline with ProRefiner
Our proposed ProRefiner contains a stack of memory-efficient global attention layers. We first represent protein structures as graphs and construct edges between nearby residues to represent their connections. In the model, attention weights are computed between every pair of residues to quantify the strength of their interactions. Different from previous works, we compute the attention weights from the residue features, the edge features and layer-specific pseudo edge features if two residues are not connected by an edge. The learnable pseudo-edge features enable the model to conduct global attention operation in an memory-efficient way. Residues can gather information from the whole protein structure even when two residues are not directly connected on the graph.
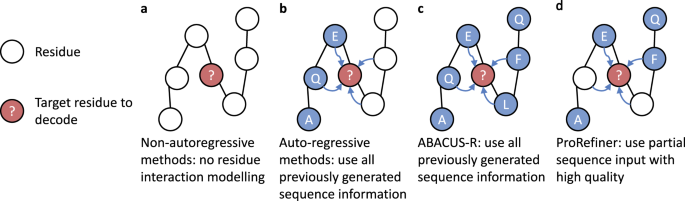
We further propose an entropy-based residue selection method to select high-quality and meaningful residue information which serves as a reference for ProRefiner's predictions. Specifically, an existing IPF model is employed to predict sequences from structures in the form of probability distributions over 20 amino acid types. Entropy of each residue's prediction is computed as a measure of model confidence, with lower entropy indicating higher confidence levels. We mask out the predictions with high entropy to reduce the noisy residue information. Then ProRefiner can complete and refine the partial residue environment and generate sequences more compatible with the corresponding 3D structure.

Learning residue interactions with ProRefiner
To assess ProRefiner's ability to learn and capture residue interactions, we analyzed the average attention weights that each residue assigns to others. We observed that many residues form important chemical bonds with those they attend to the most. Figure 2 a-c show some selected case study results, where central residues (in blue) and residues with the highest attention weights (in orange) are highlighted. For 2KCD in Figure 2 a, ProRefiner accurately identifies two hydrogen bonds: one between HIS 9 and LEU 5 on the helix, and another between ILE 70 and ASN 54 on the sheet. Similarly, in the case of T4-lysozyme (1LYD), ASP 70 forms a hydrogen bond with LEU 66 and a salt bridge with HIS 31, and both residues are among its most attended residues. ProRefiner also captures the presence of a disulfide bond between CYS 99 and CYS 94 in human Ero1-alpha (Q96HE7).
Improving protein sequence design
We conducted two sequence design tasks to evaluate the performance of ProRefiner. Firstly, we employ ProRefiner as an add-on module to refine the sequences generated by existing models, using the proposed pipeline introduced above. We experimented with multiple recent Inverse Folding models. ProRefiner demonstrates its ability to significantly refine sequence quality and improve the recovery of native sequences. Detailed results and discussions can be found in our paper. In Figure 2 d, we present the Inverse Folding results obtained on protein 2KCD. We utilized ESM-IF1 model to generate the baseline sequence, and subsequently employed ProRefiner to refine its quality. The resulting sequence can better recover the native protein structure, as assessed by Alphafold2.
Additionally, we apply ProRefiner to design single point mutants of Transposon-associated transposase B to improve its editing activity. This design scenario be seen as a special case of Inverse Protein Folding, where only one residue could be modified and the others are fixed and provided as design references. We leverage ProRefiner's predicted probabilities for the mutation site to measure the mutant stability. Amino acid types with higher probabilities are considered more stable and compatible with surrounding structure context. Following the prediction of mutant stability, we ranked the mutants accordingly and selected the top 20 mutants for experimental validation. Experiments show that 6 variants designed by ProRefiner exhibit above 1.2-fold improvement in indel activity relative to TnpB WT. Figure 2 e demonstrates the improvement of variants recommended by ProRefiner in indel activity relative to TnpB WT, as well as the indel formation at the on-target and off-target sites observed for TnpB WT and the variant with the highest activity, TnpB K84R.
Conclusion and discussion
In this work, we aim to improve the modeling and understanding of inter-body interactions within protein structures by deep learning models. While we focus on the task of structure-based sequence design, potential future research directions could involve the application of proposed model to other protein-related tasks and the examination of other biomolecules.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in