Protein purification with light – a revolution in affinity chromatography
Published in Bioengineering & Biotechnology and Biomedical Research
Proteins are the functional biomolecules of life, including enzymes, hormones, antibodies and a plethora of binding proteins, structural proteins as well as membrane proteins. To study proteins for fundamental research, or for biomedical and industrial applications, it is often crucial to obtain these biomolecules in a pure state. Conventionally, this is achieved in a laborious procedure mainly using chromatographic separation techniques, such as ion exchange chromatography (IEX), size exclusion chromatography (SEC) and the like.
This task has been considerably simplified with the invention of affinity chromatography 50 years ago. In this technique, a specific ligand or binding partner (e.g., an enzyme inhibitor or an antibody) for the protein of interest (POI) is covalently immobilized onto a stationary phase, usually a biocompatible hydrophilic polymer such as agarose. Later on, after the advent of genetic engineering, the concept of affinity tags which can easily be attached to a recombinant protein (e.g., the His-tag or the Strep-tag) has emerged.
For affinity chromatography, the POI is typically applied as a mixed solution from a cell extract or culture supernatant (i.e. containing various contaminants) in the mobile phase, thereby forming a noncovalent complex with the immobilized ligand. After thoroughly washing out the contaminants under flow with an aqueous buffer, the POI is specifically eluted in a distinct dissociation step. This can be achieved either with a solution of a competing ligand or by changing the buffer conditions of the mobile phase, for example to high or low pH, elevated salt concentration, or by applying a chemical denaturant, detergent or chelating agent.
Even though, in principle, affinity chromatography is a one-step procedure, the chemical agents applied for the elution usually need to be removed in another separation step, such as by dialysis or SEC, in order to avoid interference with subsequent investigations, for example in a cell culture assay. Consequently, we wondered if it is possible to replace the use of chemistry for the elution step by a physical driving force like temperature, electrostatics or light. While proteins are sensitive to heat and also to electrochemical reactions, they are quite resistant to electromagnetic radiation in the visible or near ultraviolet (UV) light range.
In this context, the azobenzene group, which is long known to show a switchable change in its molecular shape via light-induced cis-trans isomerisation, offered the possibility to engineer a noncovalent ligand interaction that can simply be controlled by irradiation. However, to turn this intriguing concept into reality a series of obstacles had to be overcome, which took us several years:
- Incorporation of the azobenzene group into recombinant proteins. We chose the non-canonical amino acid p-(phenyl-azo)-L-phenylalanine (Pap), which can be cotranslationally appended or inserted into a POI utilizing an expanded genetic code and amber stop codon suppression. As established methods appeared inefficient we had to optimize this technique by (i) engineering a cognate aminoacyl-tRNA synthetase (aaRS) via directed evolution, (ii) designing a one-plasmid expression vector encoding both the POI, the engineered aaRS and the cognate suppressor tRNA and (iii) engineering an Escherichia coli host strain with a less effective ribosomal release factor, RF-1. The resulting system allowed the preparation of multi-milligram quantities of various POIs carrying the Pap residue from a bacterial shake flask culture.
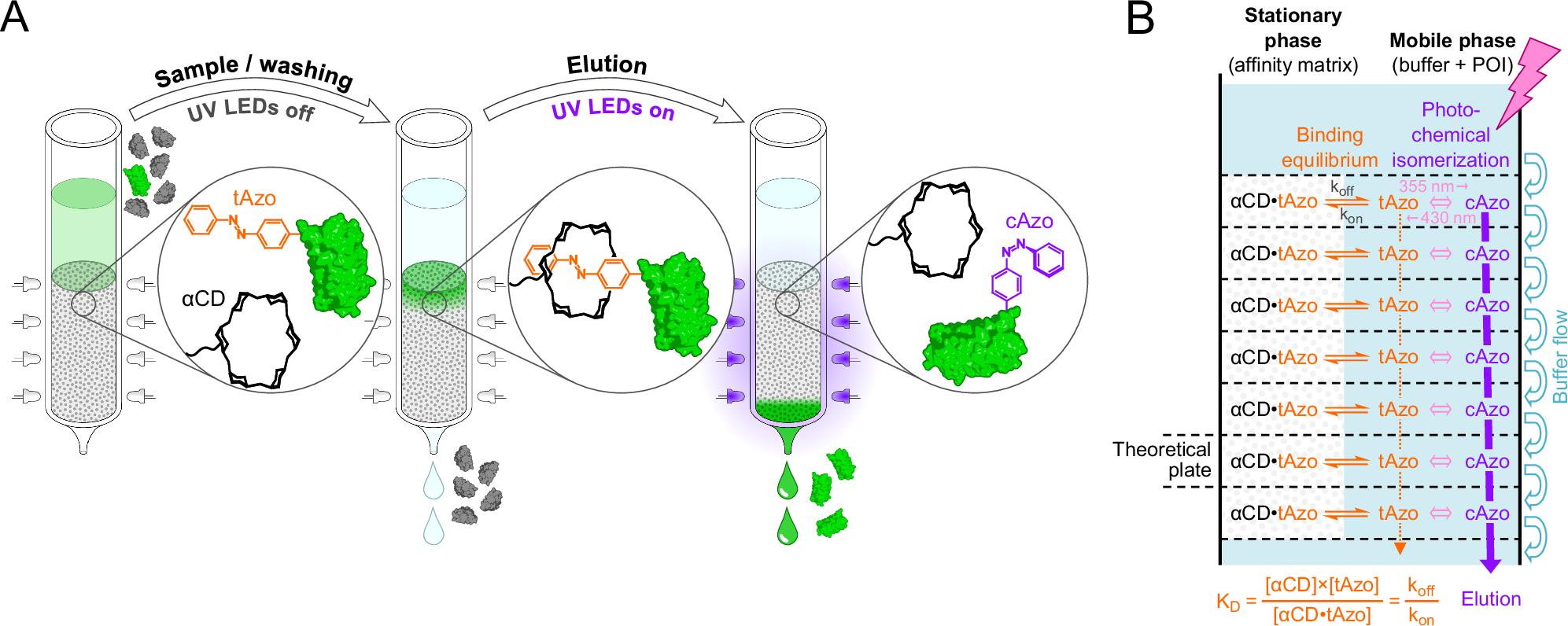
- Identification of a supramolecular interaction partner of the azobenzene side chain specifically in its trans-state. Based on the knowledge that azobenzene derivatives can form noncovalent complexes with cyclic sugar compounds, so-called cyclodextrins (CDs), we performed systematic binding studies between Pap and different commercially available CDs. Surprisingly, we found that α-CD – a ring of six glucose molecules with a hydrophobic inner and hydrophilic outer surface – forms a rather tight complex with Pap exclusively in its energetically relaxed trans-state whereas it does not interact measurably with the higher-energy and more bulky cis-configuration. This behavior was retained when Pap was incorporated at a sterically accessible position into recombinant POIs in form of the "Azo-tag" (i.e. flanked by one or two small natural amino acids and placed at the amino- or carboxy-terminus of the polypeptide chain). Furthermore, it was possible to covalently immobilize α-CD onto an established hydrophilic chromatography matrix, resulting in a stationary phase with translucent properties.
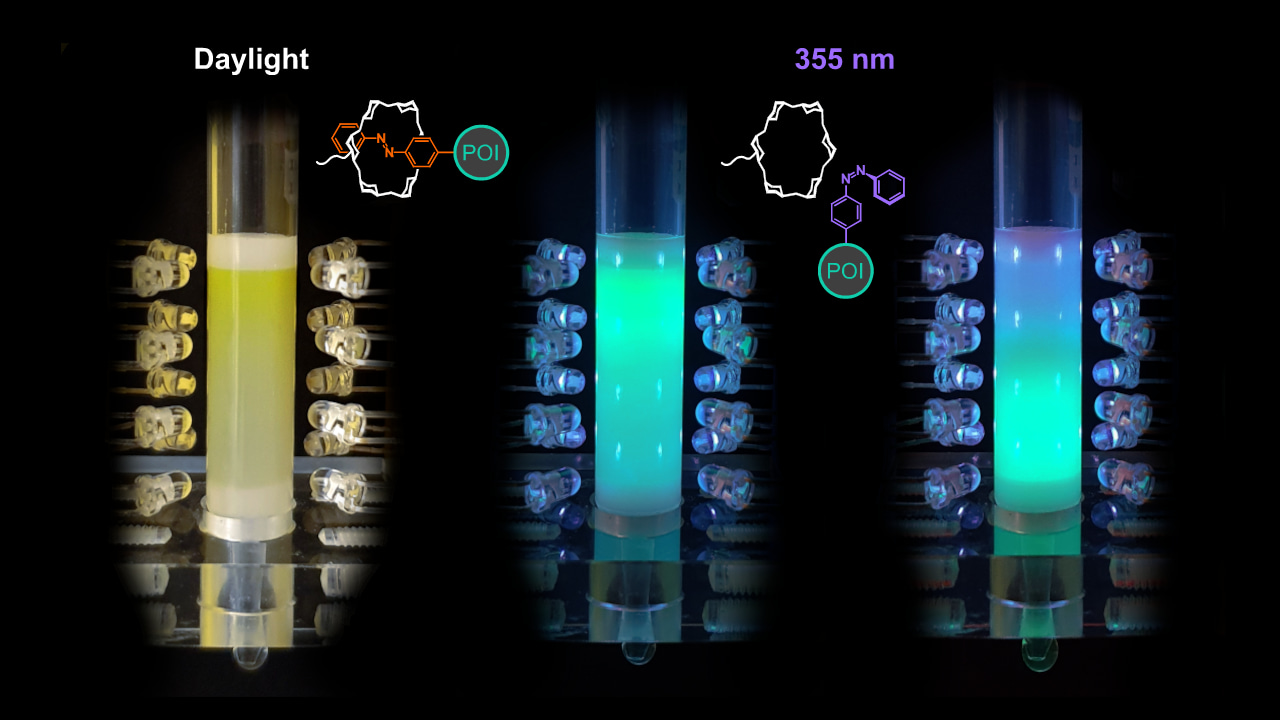
- Choice of the optimal wavelength and design of a device that allows illumination of the affinity column. The question remained if the light-switchable affinity between Pap and α-CD was suitable, considering both thermodynamic and kinetic aspects (in view of the multiple theoretical plates of the column), to control a chromatographic separation under buffer flow and if this could be achieved using an affordable experimental setup. One problem was to trigger the efficient and fast elution by quantitative switching of the azobenzene side chain into the cis-state in spite of the well-known spectroscopic overlap between the cis- and trans-isomers. Fortunately, relatively cheap LEDs have become commercially available that produce light at defined wavelengths in the near UV region, and we found out that 355 nm was optimal to achieve 95% cis-switch at a short time scale. Choice of this wavelength was significantly more effective (as determined by NMR measurements), even at a much reduced light intensity (in the low mW range), than the wavelength of 365 nm that was commonly used previously for the conversion of azobenzene compounds into the cis-state. Thus, we were able to construct a simple column housing including rows of UV LEDs to perform affinity chromatography at the laboratory scale controlled by light (see the image above provided by Peter Mayrhofer).
The resulting one-step affinity purification procedure, dubbed "Excitography", just involves application of the cellular extract containing the Azo-tagged POI, washing to remove contaminating biomolecules (including host cell proteins) under daylight or in the dark and, finally, exposure to the mild UV light in order to elute the POI in a buffer of choice, ready for subsequent experiments. Apart from the simple control of elution by a few UV LEDs, a particular advantage of this affinity chromatography is the extremely low background binding activity of the α-CD groups immobilized on the inert chromatography matrix, which allows efficient removal of all impurities during the short washing step.
Proof of concept for Excitography was initially achieved with colored and/or fluorescent proteins (e.g. GFP, see the image above) which permitted the visual assessment of the chromatography process. Subsequently, diverse other proteins were successfully purified from bacterial cell extracts with the help of the Azo-tag by light-controlled affinity chromatography, including enzymes and various immunoglobulin (Ig) fragments (e.g., scFv and dAb). Beyond that, using a tiny Azo-tagged adapter molecule based on protein G, even an intact antibody (trastuzumab) was purified from a cell culture medium, thus efficiently separating it from the large excess of albumin. Of note, the presence of the Azo-tag did not functionally hamper the POIs, as shown in a series of control experiments, while exposure to the mild UV light during the short elution step had no detrimental effect on the protein integrity either.
Thus, Excitography appears particularly useful for biomedical research on proteins in the laboratory but also has potential for applications at larger scale, if using a suitable column design that allows efficient illumination. Furthermore, benefits of the light-controlled elution of Azo-tagged POIs in a buffer of choice, devoid of any chemical contaminants as needed for conventional affinity chromatography, were demonstrated for the 96-well microtiter plate format, where the resulting protein solution can be directly applied to biochemical or cell culture assays in a high throughput format. Finally, based on the now established fundamental principles, even smarter implementations of affinity chromatography are envisioned, for example with the design of a functionalized stationary phase which itself changes its protein-binding activity in a light-controlled manner.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in