PTEN-regulated PI3K-p110 and AKT isoform plasticity controls metastatic prostate cancer progression
Published in Cancer and Cell & Molecular Biology
One of the largest challenges in prostate cancer (PC) research is identifying actionable drivers that might synergize with current inhibitors of the androgen axis, thereby preventing progression to the so-called castration-recurrent (CRPC) phase that marks the disease’s lethal phenotype. Relative to other solid tumor types, PC has a relatively low-mutational burden, with loss of PTEN representing one of the most frequent mutations. The absence of PTEN’s lipid phosphatase activity promotes the activation of the serine/threonine kinase AKT via the production of phosphatidylinositol 3-phosphate (PIP3) by PI3K, evidenced by increased relative levels of AKTpoS473. This results in increased oncogenic survival, proliferation, and metabolic pathways (1). Even though >60% of CRPC cases exhibit PTEN loss (2), pan-AKT or pan-PI3K inhibitors failed as monotherapies, and only limited benefits were shown when pan-AKT inhibitors were combined with either docetaxel (3) or the androgen axis inhibitor, abiraterone (4).
In parallel, CRPC lesions exhibit increased levels of activated Src tyrosine kinase not resulting from Src mutations (5). Src is required for androgen-independent PC cell line proliferation in vitro (6) and for CR growth in vivo (7). Nonetheless, Src inhibitors such as Dasatinib or Saracatinib, have not increased survival in CRPC patients when combined with docetaxel or abiraterone (8), even though a recent pre-clinical study suggests that Saracatinib might synergize with the androgen receptor antagonist, enzalutamide (9).
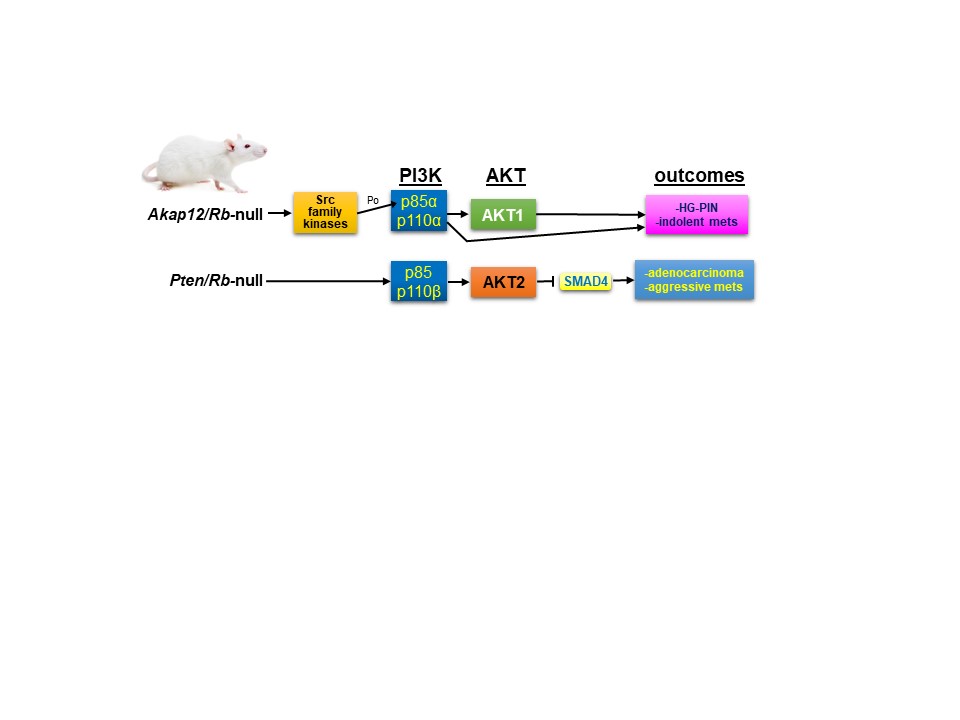
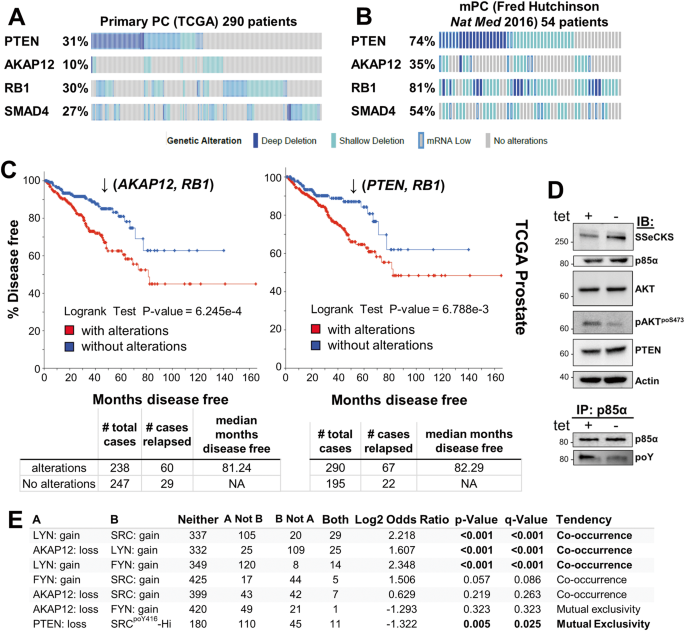
AKAP12/SSeCKS functions as a metastasis suppressor in PC (10-12) by directly scaffolding Src to lipid rafts without affecting intrinsic Src kinase activity, thereby disengaging Src from oncogenic MEK/ERK, STAT3 and PI3K/AKT signaling complexes (13). Over a third of CRPC lesions are marked by the loss of the AKAP12 gene, and furthermore, cases with PTEN loss were mutually exclusive from those with AKAP12 loss. In order to produce a genetically engineered mouse model (PC) of Src’s oncogenic role in PC, Akap12-/- mice were produced with prostate-specific loss of Rb, based on TCGA (The Cancer Gene Atlas) data showing coincident loss of AKAP12 and RB1 in CRPC. Consistent with this and the putative role of SSeCKS/AKAP12 as a metastasis suppressor, male Akap12/Rb-null mice developed high-grade intraepithelial prostatic hyperplasia (HG-PIN) as well as dissemination to lymph nodes of tumor cells expressing markers both basal and luminal epithelial linages (12). These lesions also were marked by increased Src oncogenic signaling including significant relative increases in AKTpoS473 (12,14).
In the current study (15), our group sought to understand how the loss of PTEN or AKAP12 affected the roles of PI3K and AKT as drivers of prostate cancer metastasis. We compared two mouse PC models, Pten;RbPE:-/- and Akap12-/-;RbPE:-/-, which expressed similar levels of activated AKT, yet which differed in their disease progression: whereas the former produced aggressive adenocarcinomas and systemic metastases, the latter produced HG-PIN plus indolent metastatic lymph node disseminations. We addressed whether the different disease outcomes might result from differential dependencies on PI3K and/or AKT isoforms. Indeed, there is a growing body of literature showing differential, even oppositional roles for PI3K/AKT isoforms in cancer progression (reviewed in (16)). We addressed this issue by producing human and mouse PC cell line pairs isogenic for PTEN and then using RNAi and/or isoform-specific small molecule inhibitors to determine the dependence of in vitro or in vivo parameters of metastatic growth or motility on specific PI3K-p110 or AKT isoforms. The parameters included chemotaxis, Matrigel invasiveness, colony growth in soft agar, survival in 2D (clonogenic) and 3D (anchorage-independent) growth conditions, orthotopic tumor growth and spontaneous metastasis formation. Our data showed that metastatic aggressiveness in the absence of PTEN was driven by a combination of p110β and AKT2 (and, in the case of invasiveness, AKT3). In contrast, PTEN expression not only diminished metastatic growth/motility parameters, but also shifted dependence onto p110α plus AKT1. Given evidence that our Akap12/Rb-null PC model reflected increased Src-mediated oncogenic signaling, our data further imply that PC progression is driven by two divergent drivers of PI3K/AKT signaling: PTEN loss vs. Src-family kinase activation. This is borne out in TCGA data showing little overlap between PC patients with Src, FYN or LYN gene amplifications and those with PTEN deletions.
Another of our findings is that only Pten/Rb-null tumors showed the loss of the known metastasis suppressor, SMAD4 (17). SMAD4 downregulation correlated with increased AKT2 expression in human PC cell lines, and treatment with the AKT2 inhibitor, CCT128930, resulted in increased SMAD4 levels in PTEN-negative but not in PTEN-positive PC cells lines. Of note is that SMAD4 knockdown induced increased chemotaxis.
A final finding indicates that in some PC cell lines, the ability to demonstrate suppression of AKT activation levels (relative AKTpoS473) by PTEN often requires growth of cells in 3D conditions, likely reflecting the fact that most of the growth/motility assays where AKT2 or AKT3 plays major roles involve some aspect of 3D.
There are several translational impacts of our study. The first is that inhibition of PC metastasis likely requires the combined targeting of p110β and AKT2 in PTEN deficient cases (which are the majority of CRPC cases). The fact that p110 isoform specific inhibitors (or RNAi) failed to significantly inhibit the metastatic parameters of PTEN-deficient PC cells accentuates the notion that there are AKT-independent PI3K and PI3K-indpendent AKT progression pathways. Also, if AKT2 and AKT3 are drivers of aggressive metastatic progression, this must be mediated by AKT2/3-selective substrates not shared with AKT1. Confounding this is a dearth of known AKT2- and AKT3-specific substrates. Indeed, we show both shared and unique substrates in blots of Pten/Rb-or Akap12/Rb-null tumor lysates probed with sera specific for the canonical AKT phosphorylation motif, RxxSpo/Tpo. Lastly, the failure of several so-called pan-AKT drugs, such as MK2206, in CRPC trials might be due to the fact that they are more AKT1-selective in vivo (16). With the recent development of allosteric AKT isoform-selective inhibitors by Rauh’s group (18,19) as well as AKT inhibitors that efficiently co-target all three AKT isoforms, such as Capivasertib or Vevoridertib, there is excitement that if these drugs show better clinical effects against CRPC, this will lead to a renewed focus on targeting the PI3K/AKT axis, especially in combination with androgen axis inhibitors.
Reference List
(1) Deocampo ND, Huang H, Tindall DJ. The role of PTEN in the progression and survival of prostate cancer. Minerva Endocrinol 2003;28:145-53.
(2) Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015;161:1215-28.
(3) Crabb SJ, Griffiths G, Marwood E, Dunkley D, Downs N, Martin K, et al. Pan-AKT Inhibitor Capivasertib With Docetaxel and Prednisolone in Metastatic Castration-Resistant Prostate Cancer: A Randomized, Placebo-Controlled Phase II Trial (ProCAID). J Clin Oncol 2021;39:190-201.
(4) Sweeney C, Bracarda S, Sternberg CN, Chi KN, Olmos D, Sandhu S, et al. Ipatasertib plus abiraterone and prednisolone in metastatic castration-resistant prostate cancer (IPATential150): a multicentre, randomised, double-blind, phase 3 trial. Lancet 2021;398:131-42.
(5) Drake JM, Graham NA, Lee JK, Stoyanova T, Faltermeier CM, Sud S, et al. Metastatic castration-resistant prostate cancer reveals intrapatient similarity and interpatient heterogeneity of therapeutic kinase targets. Proc Natl Acad Sci U S A 2013;110:E4762-9.
(6) Chattopadhyay I, Wang J, Qin M, Gao L, Holtz R, Vessella RL, et al. Src promotes castration-recurrent prostate cancer through androgen receptor-dependent canonical and non-canonical transcriptional signatures. Oncotarget 2016;8:10324-47.
(7) Su B, Gillard BM, Gao L, Eng KH, Gelman IH. Src controls castration recurrence of CWR22 prostate cancer xenografts. Cancer Med 2013;2:784-92.
(8) Gao L, Han B, Dong X. The Androgen Receptor and Its Crosstalk With the Src Kinase During Castrate-Resistant Prostate Cancer Progression. Front Oncol 2022;12:905398.
(9) White RE, III, Bannister M, Day A, Bergom HE, Tan VM, Hwang J, et al. Saracatinib synergizes with enzalutamide to downregulate AR activity in CRPC. Front Oncol 2023;13:1210487.
(10) Xia W, Unger P, Miller L, Nelson J, Gelman IH. The Src-suppressed C kinase substrate, SSeCKS, is a potential metastasis inhibitor in prostate cancer. Cancer Res 2001;61:5644-51.
(11) Su B, Zheng Q, Vaughan MM, Bu Y, Gelman IH. SSeCKS metastasis-suppressing activity in MatLyLu prostate cancer cells correlates with VEGF inhibition. Cancer Res 2006;66:5599-607.
(12) Ko H-K, Akakura S, Peresie J, Goodrich DW, Foster BA, Gelman IH. A transgenic model for early prostate metastasis to lymph nodes. Cancer Res 2014;74:945-53.
(13) Su B, Gao L, Meng F, Guo LW, Rothschild J, Gelman IH. Adhesion-mediated cytoskeletal remodeling is controlled by the direct scaffolding of Src from FAK complexes to lipid rafts by SSeCKS/AKAP12. Oncogene 2013;32:2016-26.
(14) Akakura S, Huang C, Nelson PJ, Foster B, Gelman IH. Loss of the SSeCKS/Gravin/AKAP12 gene results in prostatic hyperplasia. Cancer Res 2008;68:5096-103.
(15) Miller K, Degan SE, Wang Y, Cohen J, Ku SY, Goodrich DW, et al. PTEN regulated PI3K-p110 and AKT isoform plasticity controls metastatic prostate cancer progression. Oncogene 2023;online ahead of print.
(16) Degan SE, Gelman IH. Emerging Roles for AKT Isoform Preference in Cancer Progression Pathways. Mol Cancer Res 2021;19:1251-7.
(17) Ding Z, Wu CJ, Chu GC, Xiao Y, Ho D, Zhang J, et al. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature 2011;470:269-73.
(18) Quambusch L, Depta L, Landel I, Lubeck M, Kirschner T, Nabert J, et al. Cellular model system to dissect the isoform-selectivity of Akt inhibitors. Nat Commun 2021;12:5297-25512.
(19) Quambusch L, Landel I, Depta L, Weisner J, Uhlenbrock N, Müller MP, et al. Covalent-Allosteric Inhibitors to Achieve Akt Isoform-Selectivity. Angew Chem Int Ed Engl 2019;58:18823-9.
Follow the Topic
-
Oncogene
This journal aims to make substantial advances in our knowledge of processes that contribute to cancer by publishing outstanding research.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in