Putting phage to work for covalent inhibitor design
Published in Bioengineering & Biotechnology

Explore the Research

Identification of highly selective covalent inhibitors by phage display - Nature Biotechnology
Covalent inhibitors with high selectivity are rapidly identified by phage display.
My quest to understand and control the selectivity of molecules that bind proteins through irreversible covalent bonds began as a graduate student at MIT in the mid 90’s. At the time, the idea of designing drugs that irreversibly modify proteins was taboo in the pharmaceutical industry. The general thinking was that these types of molecules were dangerous because their generally reactivity could lead to undesired alterations of proteins that could result in toxicity or activation of an immune response. My view, even as a naïve graduate student, was that this was only a valid concern if a compound lacked sufficient binding selectivity. It seemed to me that molecules with overall weak chemical reactivity should be able to be equipped high affinity targeting elements such that a productive covalent bond to a target protein could only form when the molecule bound with sufficient affinity and correct orientation.
My chance to test this hypothesis came sooner than I expected when my PhD advisor, Hidde Ploegh, approached me and told me that he needed an inhibitor for the mammalian proteasome, MG-132, which was not commercially available. This tri-peptide aldehyde is an early precursor to the FDA approved proteasome inhibitor, Velcade and was originally made by a company founded by Fred Goldberg at Harvard Medical School called MyoGenics. I knew I could make the compound and I was particularly excited about the structure because I also knew its C-terminal aldehyde could be readily converted into a vinyl sulfone, a new class of reactive electrophiles that a chemist at Khepri Pharmaceuticals, Jim Palmer, had recently demonstrated was effective at covalent irreversible inhibition of cysteine proteases1. I therefore reasoned that it should be possible to convert the reversible binding MG-132 aldehyde into a covalent, irreversible vinyl sulfone probe for the proteasome in a single chemical synthesis step using the Horner-Wadsworth-Emmons reaction.
My friends working in total synthesis labs told me that I was wasting my time because the hydroxyl nucleophile would not be capable of carrying out the required Michael addition that a cysteine thiol was so good at doing. I decided that since my plan was to challenge a dogma of covalent molecule specificity, that I was not going to let a pre-conceived idea about the general reactivity of hydroxyls stop me from performing the actual experiment. So I synthesized Z-L3VS in one step from MG-132 and then synthesized a close analog, NIP-L3VS which contained a site for attachment of a 125-iodine label that would allow me to track where the molecule went inside the cell and more importantly, determine just how selective it really was (Fig. 1).

Figure 1. The process of making my first covalent probe. It started with the MG-132 aldehyde that I converted to the corresponding vinyl sulfone (Z-L3-VS) and then to a version that could be labeled with 125-Iodine (125I-NIP-L3VS). This radioactive probe specifically labeled the beta-5 subunit of the proteasome in live cells (see autoradiogram of gel at bottom right; intact US11, HOM-2 and T2 cells were labeled - image taken from my 1997 paper2).
After adding my newly synthesized radioactive proteasome probe to live cells and letting is sit for an hour, I collected and washed the cells, added sample buffer, loaded it all on an SDS-PAGE gel and exposed the gel to film. I distinctly remember standing outside the darkroom waiting for my results to roll out of the processing machine. Much to my initial chagrin, the film that emerged seemed to be completely blank, but then as the lower half of the film emerged, I immediately saw what looked to be a single intense band of radioactivity (see the gel image in Fig. 1). I later proved that this radioactive band on my gel was the beta-5 subunit of the proteasome that had been covalently labeled by my vinyl sulfone probe inside the cell2. So not only were my chemist friends wrong about the reactivity of the vinyl sulfone, I had just convinced myself that a molecule containing a reactive electrophile could possess such a high level of target selectivity, that even when presented with thousands of other possible protein targets inside the cell, it only formed a covalent bond with one. I was hooked. I knew this finding was something that would not only shape my PhD thesis but also all of my future directions in science.
From that point on, I devoted my scientific career to finding ways to develop and apply new classes of covalent binding molecules. I went on to start my own lab as a Sandler Faculty Fellow at UCSF in late 1998, along with the two other fellows at the time, Joe DeRisi and Anita Sil. While there, I expanded the use of covalent inhibitors to the lysosomal cysteine proteases3,4 and several cysteine proteases in parasite pathogens5. At about the same time, Ben Cravatt was building up his program as an Assistant Professor at Scripps. He made a trip to UCSF to give a seminar and as fate had it, I ended up on his schedule. That meeting ended up being one the most memorable discussions of my career. We talked at length about our interest in chemical probes and the potential of these agents for use in studying the basic biology of enzymes. Ben had recently synthesized his now famous FP-biotin probe to study the serine hydrolase, fatty acid amide hydrolase (FAAH)6 and there were a number of striking similarities between his thinking and mine. He agreed to send me his FP probe and we decided to collaborate (Fig. 2). To this day, we are friends and colleagues and I consult him when I have interesting ideas and want his input. While we had not yet fully realized the power of our probes back then, we had already laid the groundwork for the field of activity-based protein profiling (ABPP).

Figure 2. Original letter from Ben Cravatt in early 2000 after our meeting at UCSF when he sent me a sample of his FP-biotin probe to test in my laboratory.
One of the things I quickly learned in the early days of building and applying activity-based probes (ABPs) was that the most important parameter to understand and regulate is selectivity. Many of the first ABPs that I made, including the peptide vinyl sulfones for the proteasome, were in fact weak inhibitors of the target enzyme. What made them interesting and useful as ABPs was this weak reactivity made them able to bind a target with an exceptionally high degree of selectivity. This selectivity could manifest itself at the level of a single enzyme, as in the proteasome2 or at the enzyme family level, as in the cathepsins3,4 or serine hydrolases7,8. While Ben worked to expand the use of broad-spectrum ABPs as ‘chemical proteomic’ tools, I was most intrigued about the potential to make probes that could label a single protease of interest, even in the context of a whole organism, such as a mouse. If it was possible to make reagents with such a high degree of selectivity, it would be possible to track the activity of a specific enzyme in cells and tissues during disease progression or to assess drug target engagement in vivo.
In late 2001, after three years at UCSF, I made what some people considered a bold decision to leave my position in academia and go to Celera Genomics, a company that had just completed sequencing of the human genome and now was making a shift to drug development. As a surprising coincidence, one of the senior chemistry directors at Celera was none other than Jim Palmer, the very same chemist whose original paper on the vinyl sulfone electrophile had sparked my interest as a graduate student. As a Group Leader there, I focused on applying ABPs to validate specific protease drug targets and to track the pharmacodynamic (PD) properties of lead drug molecules9. Although more than a decade after my first exciting result with the covalent proteasome probe, there remained a significant stigma in pharma against developing these types of molecules as drugs. So, I was forced to pitch my group’s efforts as ‘tool development’ rather than drug development. Interestingly, one of the projects in my group was to make covalent probes for kinases that could be used for PD and target engagement studies. In a rather ironic twist of fate, a chemist in my group, Zhengying Pan (who moved to Jim Palmer’s group when I left Celera to go to Stanford) made a probe for Bruton's Tyrosine Kinase (BTK) that ended up being purchased by a biotech company called Pharmacyclics when Celera decided to end all of its drug development programs in 2006. Pharmacyclics took that ‘tool compound’ and advanced it into the clinic where it was eventually approved by the FDA and became the highly effective drug, Ibrutinib, for the treatment of B-cell cancers. This drug was the first approved covalent kinase inhibitor and its great success has led to a renewed perspective about the utility and promise for covalent inhibitors as drugs. In fact, a review of current advances in covalent drug development was published just last week in the November 9th issue of Chemical and Engineering News.
So right about now you may be reading this blog post and wondering “what does this all have to do with phage?” That is where the most recent work from my laboratory comes in. Our publication in Nature Biotechnology describes the culmination of a project that started as an idea 8 years ago at a meeting during a department seminar, much like the one I had with Ben Cravatt back in 1999. I am particularly excited about this publication because it represents an exciting new direction in inhibitor design that addresses the issue that I still believe is of greatest importance for covalent inhibitors: target selectivity.
It all started when I was invited to give a seminar at the EPFL in Switzerland in 2011 and was lucky enough to end up having Christian Heinis on my schedule. Christian had recently completed his postdoctoral studies with Sir Greg Winter at the MRC in Cambridge where he had developed a new approach to chemically modify phage displayed peptides. This approach was particularly ingenious because it allowed diversification of phage displayed peptides using a linker that induced formation of bi-cyclic peptide structures that could then be iteratively screened for binding to a protein target (Fig. 3).
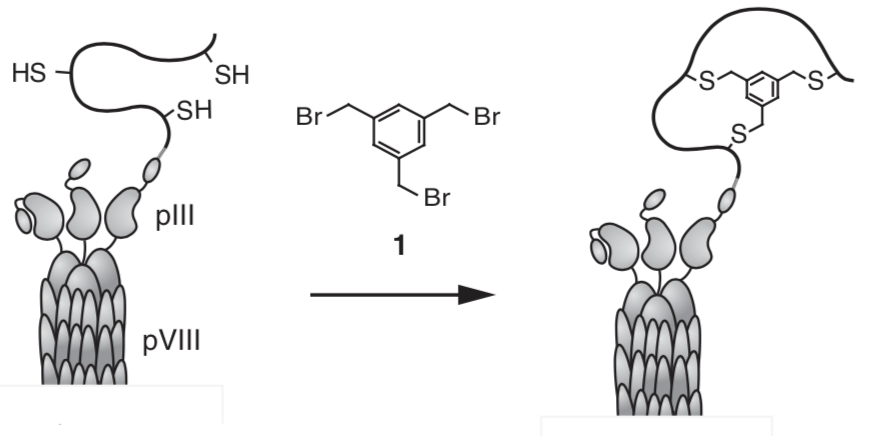
Figure 3. The original concept from Heinis to chemically cyclize peptides on the phage surface. This image is taken from his 2009 paper10 that first described the use of phage to screen bicyclic peptides for highly selective binding to protein targets.
Christian had chosen to use a protease, human plasma kallikrein (KLK), as one of his first targets and showed that by screening millions of bicyclic peptides on the phage, he could identify potent inhibitors of this protease with exceptional specificity10. In fact, I remember him showing me the data for one of his bi-cyclic peptides in which he compared potency for the human and mouse forms of KLK and how he was able to achieve a high degree of selectivity even though the two enzymes shared more than 80% sequence identity! For me this was shocking and I suddenly realized that this was the technical method that I was looking for all these years to make ABPs with the kind of near absolute specificity required for applications such as in vivo imaging. I went home to my hotel that night and though about ways to use Christian’s chemically modified phage approach for engineering covalent binding inhibitors and probes. I reasoned that because the method already involves a chemical linker to make the peptide cycles, it should be possible to simply attach a weakly reactive electrophile to the linker and kill two birds with one stone; make the cyclic peptides while simultaneously introducing the reactive ‘warhead’ for display to an active site nucleophile. This would allow panning of the resulting chemically modified phage library against a target to isolate sequences that displayed the electrophile in such a way that it would covalently bind to the target. I was so excited about this idea that I immediately contacted Christian before I got on the plane back to CA and asked him if I could come to his lab to learn the phage display method over a short sabbatical. He agreed and I headed back to Switzerland in the summer of 2012.
During this sabbatical, I returned to laboratory bench work with the help of one of Christian’s highly talented Ph.D. students, Shiyu Chen (who you will notice is the primary author of our paper). Shiyu taught me how to grow, titer, chemically modify and screen phage libraries. While I was only in the lab for 3 months, I learned the techniques of phage display and solved some early issues with linker chemistry. Unfortunately, I was not able to actually validate my overall idea so I returned to Stanford with a renewed focus and plans to continue the project in my own lab. Of course, things never go as planned and the project was slowed by my inability to convince anyone in the lab to pick it up, I think because my students and postdocs thought it was one of those crazy ideas that PIs tend to kick around the lab, with little chance for success. Luckily for me, Shiyu was completing his thesis work and I was able to convince him to come to my lab at Stanford as a postdoc. He arrived in early 2015 and then picked up working on the phage project.
Over the past five years, this project has had its ups and downs. We found that the original plan to make bicyclic peptides was problematic due to altered reactivity of the linker when we added the reactive electrophile and the potential to form many positional isomers upon cyclization. We also initially intended to screen for selective covalent inhibitors of the lysosomal cysteine cathepsins and later found that those proteases shave off surface proteins on the phage leading to loss of infectivity. We then took a step back and decided to simplify things by using a dichloroacetone functional group to make cyclic peptides on the phage and attach the electrophile via a hydrazone linkage to the ketone. This method worked well and allowed us to finally get effective screens going. We also chose to use the TEV protease as an initial target because there were no reported selective inhibitors of this protease and it has a high selectivity for a well-defined linear peptide sequence in its substrates. As you will hopefully see from reading the paper, we eventually got the method working robustly and were able to identify highly selective covalent inhibitors of TEV protease that displayed amino acid residues in the cyclic peptide region in a manner that mimics the native substrate. You may also notice that these covalent TEV inhibitors make use of my favorite electrophile from the start of this story, the vinyl sulfone. I found myself having another one of those exciting moments that you always remember in your scientific career when Shiyu showed me inhibition data for the phage-selected cyclic peptide vinyl sulfone inhibitor in which he compared its activity for TEV with its activity against two lysosomal cysteine cathepsins (Fig. 4). Even though cathepsins are effectively inhibited by linear peptide vinyl sulfones and the cyclic TEV inhibitor contains residues that are optimal for cathepsins, we saw absolutely no inhibition of these highly active off-targets, even when using high micromolar concentrations of the inhibitor. This was the kind of near absolute specificity that I had seen in Christian’s office back in 2011 and something that I now firmly believe is possible to achieve for covalent binding molecules.
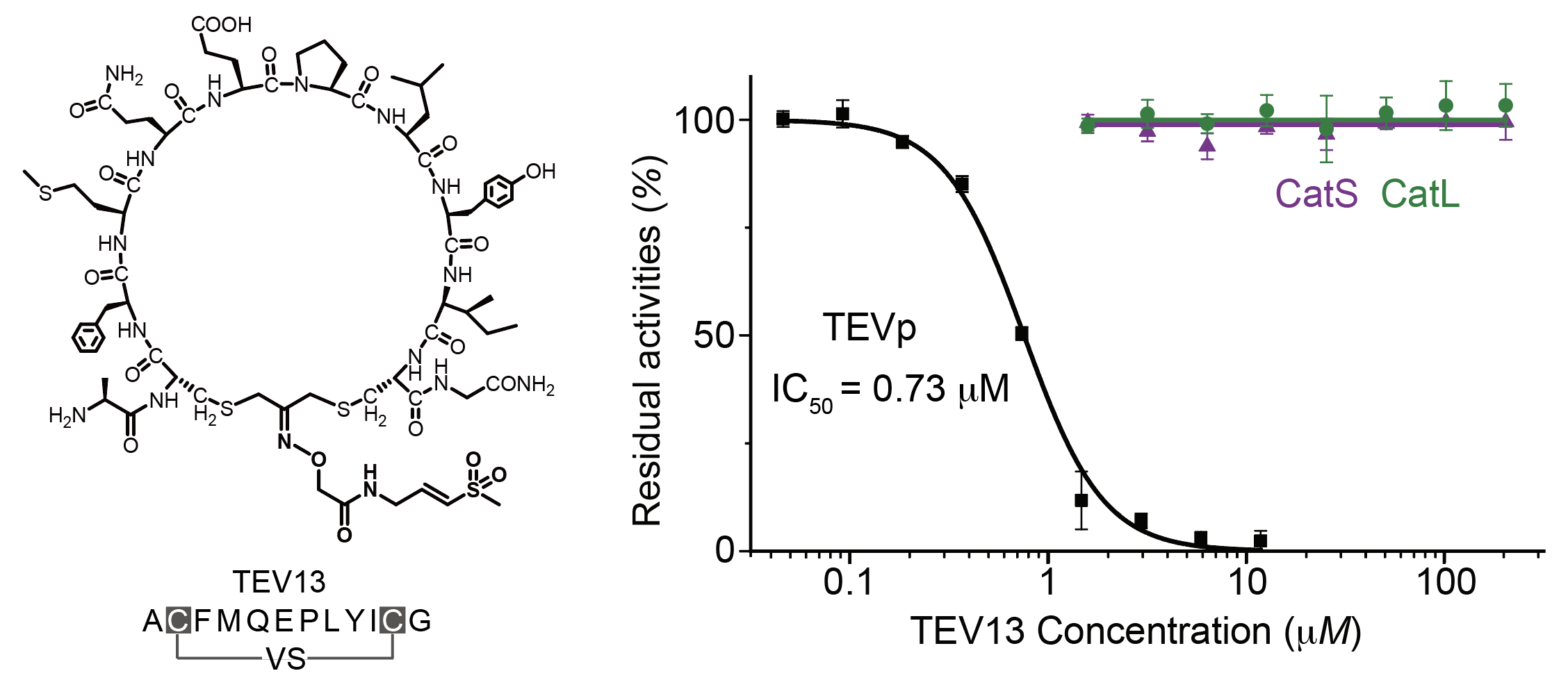
Figure 4. The high selectivity of the phage-selected cyclic peptide vinyl sulfone TEV13. The graph at right shows the dose response profile of the inhibitor for TEV relative to the two highly active cysteine cathepsins S and L which have broad substrate specificity and which are potently inhibited by linear peptide vinyl sulfones.
To further confirm that the approach could be broadly applied, we went on to test the phage method for a serine hydrolase target using a different class of electrophile, the diphenylphosphonate. We chose a serine hydrolase target, FphF, that is part of a family of enzymes that we had recently identified in Staphylococcus aureus that play important roles in establishing infection inside a host11. This was an optimal choice of targets because we could assess selectivity of the resulting covalent probes using members of this family that have highly similar serine hydrolase domains. The resulting cyclic peptide probe for FphF, like the TEV selected probe, was both potent and exceptionally selective. It showed virtually no activity against the highly related FphB hydrolase, providing further validation that the phage display approach can be generally used to control the selectivity of weak electrophiles by identifying highly selective targeting elements.
In many ways this paper, which was published close to 25 years after my first exciting result with an 125-labeled peptide vinyl sulfone, is a major milestone for my group’s efforts to demonstrate the power of covalent binding ligands. It provides a potentially general way to get phage to do all the difficult and time-consuming medicinal chemistry work to find highly selective covalent binders. We are extremely excited about this technology and are currently moving ahead full-speed on a number of other target applications. I believe that this approach will yield many new and useful molecules that in addition to being 'tools' may also become new classes of drugs that zero in on their targets with exquisite specificity and don’t let go.
- Palmer, J.T., Rasnick, D., Klaus, J.L. & Bromme, D. Vinyl sulfones as mechanism-based cysteine protease inhibitors. J Med Chem 38, 3193-3196 (1995).
- Bogyo, M. et al. Covalent modification of the active site threonine of proteasomal beta subunits and the Escherichia coli homolog HslV by a new class of inhibitors. Proc Natl Acad Sci U S A 94, 6629-6634 (1997).
- Bogyo, M., Verhelst, S., Bellingard-Dubouchaud, V., Toba, S. & Greenbaum, D. Selective targeting of lysosomal cysteine proteases with radiolabeled electrophilic substrate analogs. Chem Biol 7, 27-38 (2000).
- Greenbaum, D., Medzihradszky, K.F., Burlingame, A. & Bogyo, M. Epoxide electrophiles as activity-dependent cysteine protease profiling and discovery tools. Chem Biol 7, 569-581 (2000).
- Greenbaum, D.C. et al. A role for the protease falcipain 1 in host cell invasion by the human malaria parasite. Science 298, 2002-2006 (2002).
- Liu, Y., Patricelli, M.P. & Cravatt, B.F. Activity-based protein profiling: the serine hydrolases. Proc Natl Acad Sci U S A 96, 14694-14699 (1999).
- Jessani, N., Liu, Y., Humphrey, M. & Cravatt, B.F. Enzyme activity profiles of the secreted and membrane proteome that depict cancer cell invasiveness. Proc Natl Acad Sci U S A 99, 10335-10340 (2002).
- Jessani, N. et al. Carcinoma and stromal enzyme activity profiles associated with breast tumor growth in vivo. Proc Natl Acad Sci U S A 101, 13756-13761 (2004).
- Pan, Z. et al. Development of activity-based probes for trypsin-family serine proteases. Bioorg Med Chem Lett 16, 2882-2885 (2006).
- Heinis, C., Rutherford, T., Freund, S. & Winter, G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat Chem Biol 5, 502-507 (2009).
- Lentz, C.S. et al. Identification of a S. aureus virulence factor by activity-based protein profiling (ABPP). Nat Chem Biol 14, 609-617 (2018).
Follow the Topic
-
Nature Biotechnology

A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in