Quantifying how Single Dose Ad26.COV2.S Vaccine Efficacy Depends on Spike Sequence Features
Published in Protocols & Methods, Biomedical Research, and General & Internal Medicine
Let’s set the epidemiological Wayback machine to early 2021. The COVID outbreak was at its greatest peak yet, and following an unprecedented international effort, led by the U.S. Government COVID-19 Response Team, the Moderna and Pfizer-BioNTech vaccines had just received emergency use authorization in the United States and were on a limited roll out to medical personnel and essential workers.
At the same time, the SARS-CoV-2 virus was showing its crafty capacity for adaptation, and the original outbreak lineage was making way for emerging variants of concern. The United States was serving as a proving ground for minor breakaway strains with geocentric names like the “New York variant” and the “California variant”, while South Africa was awash in Beta, and Latin America hosted a cage match with a deep roster of Greek-lettered lineages that fought for supremacy. The Summer of Delta was right around the corner, and Omicron’s long dark winter was still a glimmer in Pestilence’s eye.
Meanwhile, another vaccine trial was underway: the ENSEMBLE randomized, placebo-controlled phase 3 trial (NCT04505722), evaluating the efficacy of Janssen’s single-dose Ad26.COV2.S vaccine. This was the most extensive efficacy trial of its time, with over 40,000 participants enrolled across South Africa and seven countries in the Americas, including the United States. At the trial’s conclusion, the vaccine efficacy of Ad26.COV2.S was found to be 56% against symptomatic COVID-19, and it soon received emergency use authorization in the United States.
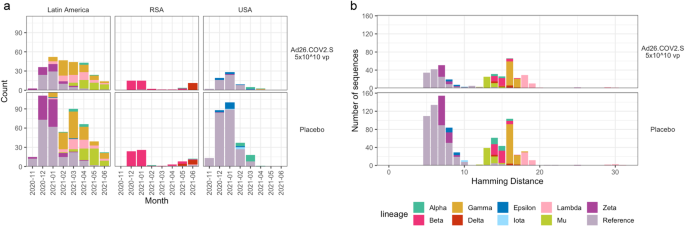
In this trial, of the 1,545 participants who were diagnosed with COVID, we were able to sequence full genomes of the SARS-CoV-2 virus from 1,224 of them (378 vaccine recipients, 846 placebo recipients). From these, we compared the Spike proteins (as amino acid sequences) between those participants who received the vaccine and those who received the placebo. This type of analysis is referred to as a “sieve analysis”: the vaccine serves as a metaphorical kitchen sieve, preventing some viruses from establishing a symptomatic infection, while others manage to sneak through. Because this was a randomized trial, the results of such an analysis can be interpreted as how vaccine made an impact on the genetic composition of the infecting viruses.
During the time of this trial, the diversity of SARS-CoV-2 was relatively low in both the United States and South Africa. The U.S. was still in the early phases of the pandemic: while having 416 COVID cases in the trial, nearly 85% of all sequenced viruses consisted of the “reference” ancestral lineage (by this I mean the original outbreak lineage B.1 with the D614G mutation). South Africa showed a similar homogeneity: while this study population was relatively small, of the 125 cases (of 172 total) with a sequenced virus, 95 (76%) of them were of the Beta variant.
Among the six Latin American countries (Argentina, Brazil, Chile, Colombia, Mexico, and Peru), however, it was a different situation altogether. In the trial, 963 cases occurred in these countries, and we were able to obtain sequences from 776 of them. These sequenced viruses were from a diverse Greek alphabet soup that included Alpha (1.8%), Epsilon (0.3%), Gamma (23.7%), Lambda (11.3%), Mu (12.2%), Zeta (16.1%) and the reference ancestral lineage (34.6%).
Such a high level of viral diversity made it more likely that any difference we found would be scientifically relevant, and while we looked at these three geographic regions separately and pooled together, our analysis focused on the Latin American population for this reason.
Our strongest finding is that the vaccine performed the worst against the Lambda variant [vaccine efficacy = 10.8% (95% C.I.: -34.6% - 41.0%)], leading us to characterize Lambda as an escape variant. Originally known as the PANGO lineage C.37, the Lambda variant was initially observed in Peru and, while it faded away within the year, it did feature a number of novel mutations that appeared in later (and even current) variants, as an example of convergent evolution and serving as a harbinger of variants to come.
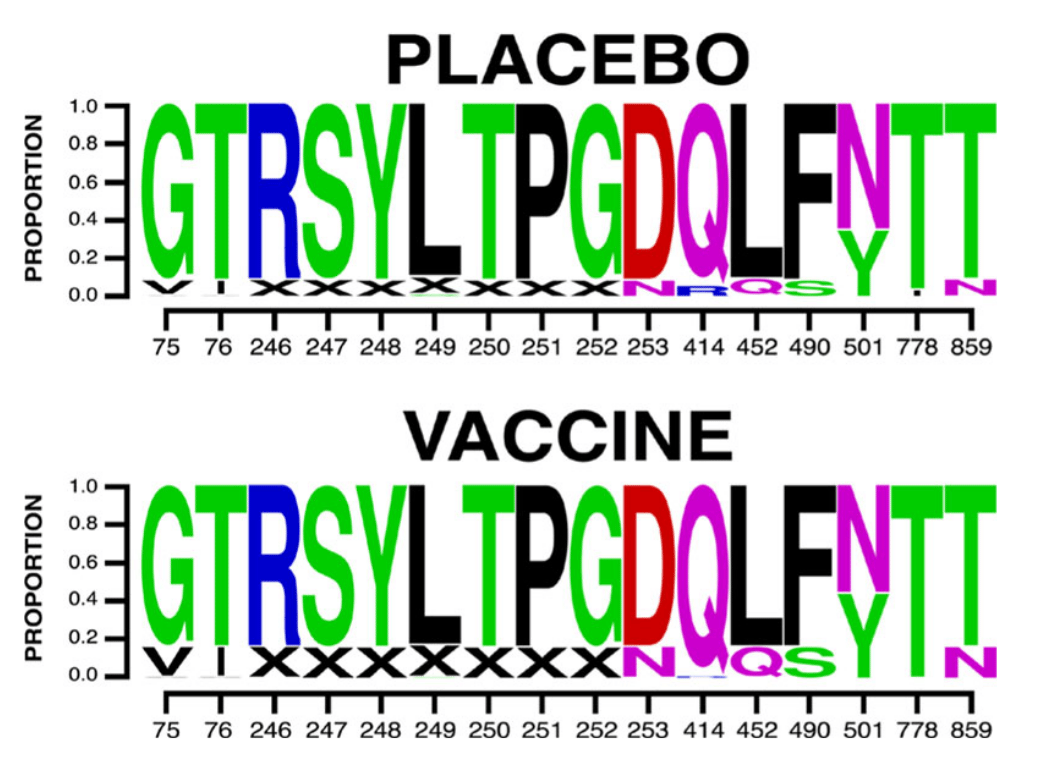
Figure 1: A sequence logo of the mutations found to affect vaccine efficacy among cases in the ENSEMBLE trial. "X" indicates a gap. Adapted from Figure 2e in the paper.
Among these mutations associated with vaccine efficacy are ten sites in the N-terminal domain (NTD) (75, 76, 246, 247, 248, 249, 250, 251, 252 and 253) and four in the receptor binding domain (RBD) (positions 414, 452, 490 and 501). Mutations at three of these sites (252, 490 and 501) are present in currently circulating Omicron sub-lineages and recombinants, and six of them (76, 247, 248, 250, 251 and 452) appeared in prominent lineages that have since fizzed out. Of special note is the F490S mutation: the vaccine worked best against the wild type F490 virus, and efficacy suffered if the phenylalanine was substituted with a serine. While rare until the end of 2022, this F490S mutation became dominant in early 2023 with the emergence of the XBB.1.5 recombinant lineage.
We employed a number of unique approaches to identify sequence differences that affected vaccine efficacy among COVID cases, including:
-
Differences in individual amino acid residues compared to the vaccine;
-
Partial- or whole-sequence similarity to the vaccine;
-
Two types of antibody escape scores;
-
Mutation patterns in site clusters in the NTD supersite epitope; and
-
Variable importance in machine learning predictors.
We also looked for differences in the virus defined by their sensitivity to neutralization by vaccine recipient sera. When the results from all of these methods are collated and compared, they tend to indicate the same thing: sieve effects that are linked to the Lambda variant. All roads lead to Lambda.
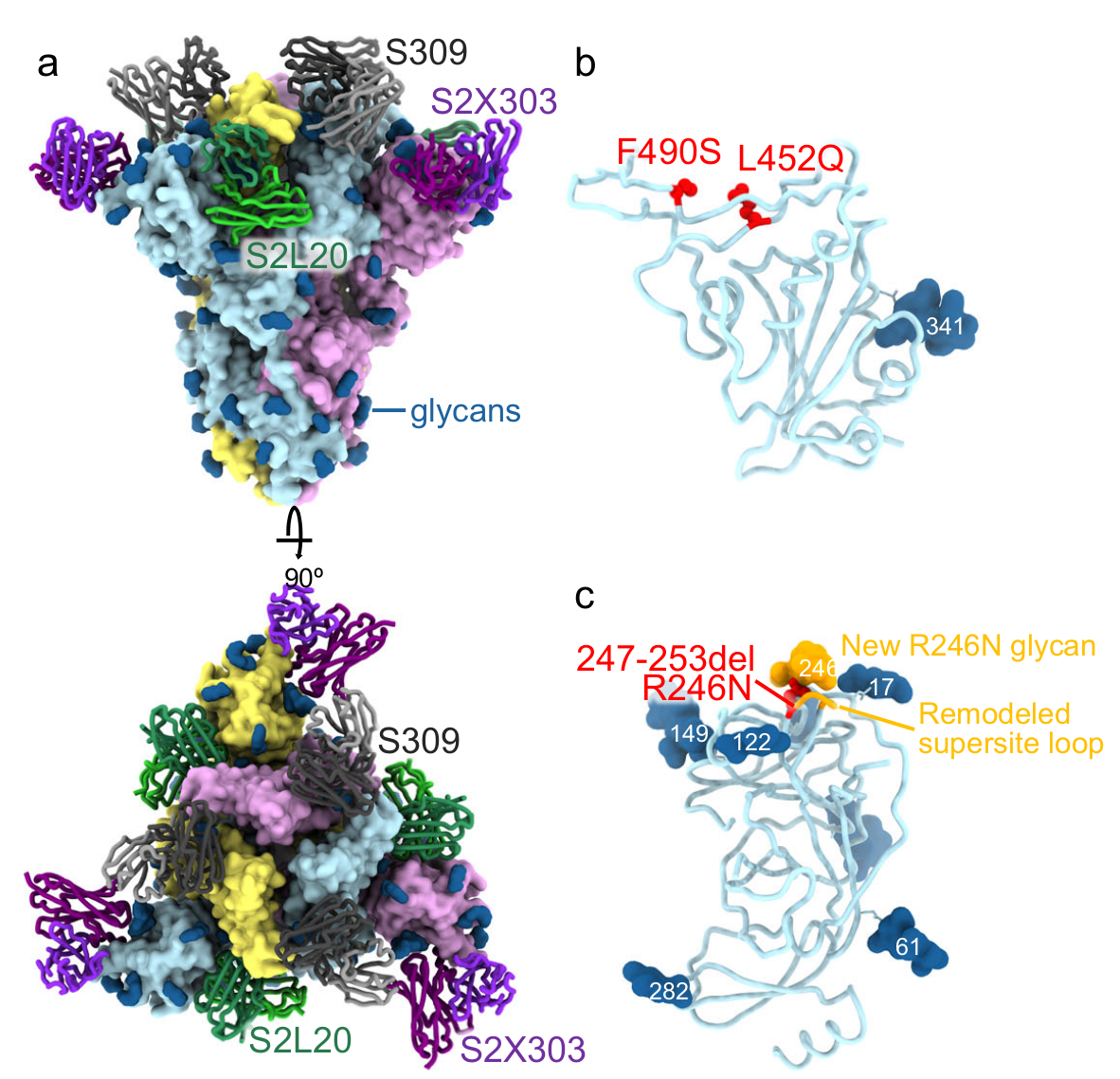
Figure 2: A CryoEM structure of the SARS-CoV-2 Lambda Spike trimer, with analysis of monoclonal antibody and ACE2 binding. (a) shows the trimer bound to the S2L20 (green), S2X303 (purple) and S309 (orange) Fabs (fragment antigen-binding). (b) is a zoomed-in view of the S309-bound Lambda RBD, showing the L452Q and F490S mutations. (c) is a zoomed-in view of the S2L20- and S2X303-bound Lambda NTD with the R246N mutation. The remodeled loop caused by the 247–253 deletion as well as the resulting R246N glycan are shown in orange. Adapted from Figure 8 in the paper.
Among our findings, however, are some results that can be interpreted generally. Most notably, efficacy was found to be highest among viruses that were most similar to the vaccine strain. As the virus gets increasingly dissimilar to the vaccine, the efficacy wanes to zero. As such, this similarity can be regarded as a biomarker of vaccine efficacy, which can have several public health applications, including guiding the selection of which strains to use with next-generation vaccines, and modeling the efficacy of a vaccine against circulating viral strains.
All of the results described above pertain to COVID cases of any disease severity level, ranging from moderate to severe or critical, even death. Our analysis also looked specifically severe-critical cases only, which was restricted to folks who experienced any of a spectrum of severe outcomes, including admission to an intensive care unit, respiratory failure or death.
And that brings us to the final takeaway point from our analysis, which is that we did not find any strong results that were specific to those who came down with severe-critical COVID. We did observe the same general effect as mentioned above (that vaccine efficacy has a moderate association with the virus’s similarity to the vaccine, which appears to be focused in the NTD), and we noticed that efficacy was dependent on some specific mutation patterns in the NTD, but none of these results passed our most-stringent criterion for statistical significance with multiple testing.
This suggests that the vaccine-linked viral mutations observed in this study mostly resulted in improved immune escape for the virus, but did not increase the disease severity in the human host.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Biosensing
Publishing Model: Hybrid
Deadline: Sep 30, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in