Quantitative Hallmarks of Leukemia
Published in Bioengineering & Biotechnology
When a patient with symptoms of leukemia enters a clinic, the diagnosis can range from “nothing to worry about” to fatal. Acute myelogenous leukemia (AML) skews towards the latter. The prognosis is bleak: only 28% of AML patients survive beyond five years.1 A cancer so heterogeneous it is described as a collection of diseases, the same genetic mutations in AML can yield vastly divergent responses to therapy. Furthermore, risk factors for AML are often uncontrollable, e.g., a prior malignancy, being male, radiation exposure and age.

Biomed. Eng., 2019)
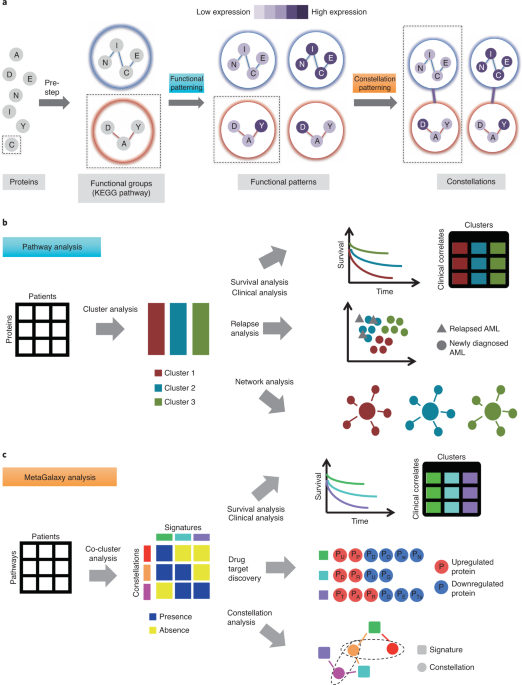
Biotechnology has shed light on AML’s complexity: cytogenetic profiling refined the World Health Organization’s classification of the disease,2 DNA methylation analysis3 and metabolic profiling4 have characterized how AML alters the differentiation of blood cells, and single cell classification of leukemia blast cells and bone marrow has enabled a greater understanding of AML’s diversity.5-7 However, a large gap remains between molecular understanding of the cancer, actionable therapeutic targets, and patient outcomes. To bridge this gap, we developed a computational framework that unravels the complexities of AML through proteomics (Figure 1).8 Applying MetaGalaxy to hundreds of patients, we identified hallmarks that relate to patient survival and remission duration (LeukemiaAtlas.org).

The Leukemia Proteome Atlas was enabled by foresight and a little fortune, both bad and good. As a medical resident in the late 1980s, Steven M. Kornblau, MD, of The University of Texas MD Anderson Cancer Center began banking tissue samples for patients, recognizing patient-derived material might be analyzed by future technologies. Once Steve invested in one of the first reverse phase protein array machines (with Gordon Mills, MD, PhD), he began profiling the AML patient biopsies. Decades after he started what became MD Anderson Cancer Center’s Leukemia Sample Bank, Steve and I met at a conference in the Colorado mountains the first winter of my independent career. Meeting Steve gave me a deep appreciation for clinical research, including its challenges.
Clinical and technical constraints made the analysis of the AML proteomic data formidable. Leukemia bone marrow biopsies are painful procedures taken infrequently, often only at initial diagnosis or relapse. This limited the data to static, one-shot or two-shot samples per patient, unlike time series proteomic datasets available for other cancers and cell lines9. We also learned ways that tissue processing and storage alter protein expression, prompting us to separate the original dataset of 511 patients’ biopsies into fresh and frozen samples for independent analysis.
The challenge was analogous to navigating the Milky Way after observing light emitted by a collection of stars, seen from locations around the world – with variable cloud coverage! 88 constellations of stars have been formally recognized by the International Astronomical Union, with each constellation subsequently assigned to one of 8 families10. We identified 11 constellations of protein functional groups and 13 signatures classifying 205 cases of acute myelogenous leukemia (Figure 2).
The MetaGalaxy approach, spearheaded and implemented by a talented graduate student Chenyue Wendy Hu (now at Uber Technologies), and designed over years of conversations between Steve, Wendy and myself along with an increasing list of integral collaborators (LeukemiaAtlas.org/team), provides a succinct set of ‘rules’ that guide the interpretation of protein signatures, and enables both local and global classification of omics screens. For AML, the method offers a powerful way to classify patients. A strength in the approach is that we sought first to identify patterns in the proteins, without regards to identifying patient outcomes. The prognostic ability of the functional groups and constellations was an unbiased outcome. This is in contrast to an international crowd-sourced data challenge we held on a subset of patient samples, where the goal was to improve predictions of clinical outcome.11 Both approaches have merits. What distinguishes MetaGalaxy from alternative machine learning methods including our own is that MetaGalaxy (1) incorporates known biology, and (2) provides a classification for patients, a succinct global decision tree (where every node is a network) and defined functional networks that inform cancer researchers as to how proteins relate to each other in AML, i.e., a mathematical framework to express hallmarks of cancer.

Biomedical engineering fosters polyglots, which enables the expertise needed to bridge computational research, laboratory work, and clinic. We (a “we” growing into dozens of engineers, scientists and oncologists) learned each other’s languages (clinical context, cancer biology acronyms, R & R shiny, medical terminology, JavaScript, engineering parameters, statistical notations, Python, etc). It is with gratitude for this constellation of dedicated individuals, and the larger galaxy of mentors, lab members, and clinical collaborators worldwide, that we provide the computational tools and atlas online. In doing so, we offer a method that can be applied broadly to interpret omics datasets and a growing compendium of proteomics for adult and childhood leukemias (Figure 3). The impetus for the Leukemia Proteome Atlas being that whether it is our team or another who translates this work to the clinic first, the insights gleaned by the MetaGalaxy analysis can make an impact for leukemia patients near-team, and an immediate impact in how preclinical drug screening is performed.

References
- Survival Statistics, SEER 18 2009-2015, National Cancer Institute (2019).
- Arber, D.A. et al., The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127, 2391-2405 (2016).
- Takahashi, K. et al., Integrative genomic analysis of adult mixed phenotype acute leukemia delineates lineage associated molecular subtypes. Nat. Commun. 9, 2670 (2018).
- Fenouille, N. et al., The creatine kinase pathway is a metabolic vulnerability in EVI1-positive acute myeloid leukemia. Nat. Med. 3, 301-313 (2017).
- Paguirigan, A.L. et al., Single-cell genotyping demonstrates complex clonal diversity in acute myeloid leukemia. Sci. Transl. Med. 7, 281 (2015).
- Van Galen, P., et al., Single-cell RNA-seq reveals AML hierarchies relevant to disease progression and immunity. Cell 176, 1265 -1281 (2019).
- Levine, J.H., et al., Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell 162, 184-197 (2015).
- Hu, C.W., et al., A quantitative analysis of heterogeneities and hallmarks in acute myelogenous leukaemia. Nat. Biomed. Eng. in press (2019).
- Hill, S.M., et al., Inferring causal molecular networks: empirical assessment through a community-based effort. Nat. Methods. 4, 310-318 (2016).
- Menzel, D.H., A field guide to the stars and planets. (Viking Press, 1982)
- Noren, D.P., et al., A crowdsourcing approach to developing and assessing prediction algorithms for AML prognosis. PLOS Comput. Biol. 12, e1004890 (2016).
Follow the Topic
-
Nature Biomedical Engineering
This journal aspires to become the most prominent publishing venue in biomedical engineering by bringing together the most important advances in the discipline, enhancing their visibility, and providing overviews of the state of the art in each field.

Related Collections
With Collections, you can get published faster and increase your visibility.
Neuroengineering
Publishing Model: Hybrid
Deadline: Oct 31, 2026
Implantable wireless communication technologies
Publishing Model: Hybrid
Deadline: Nov 28, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in