RAGE drives cell plasticity in metastatic TNBC
Published in Cancer
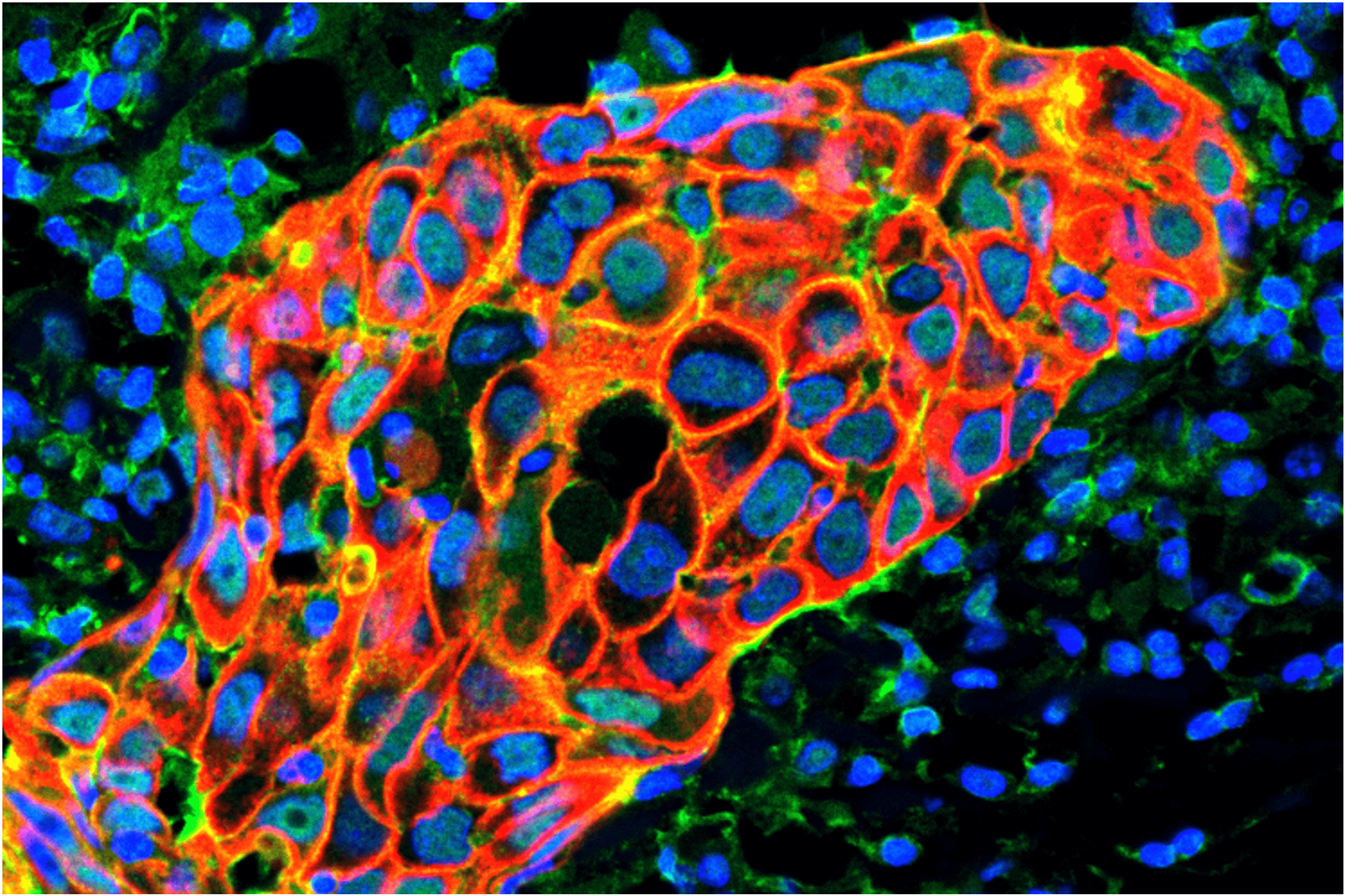
Breast cancer (BC) is the most frequent cancer among women, affecting 2 million women worldwide each year. Triple-Negative Breast Cancer (TNBC) represents about 15% of BC cases, is particularly aggressive and has a higher metastasis incidence than other BC subtypes. A group of TNBC tumors are classified as mesenchymal and they present the highest frequency of distant metastases, and the poorer prognosis among TNBC patients. The main therapeutic option for these patients is cytotoxic chemotherapy, which is largely ineffective, since a large fraction of them do not reach a pathological complete response (pCR).
The mesenchymal phenotype in tumor cells of epithelial origin can be favored by genetic mutations but accumulating evidence suggests that tumor cell plasticity is driven by external microenvironmental cues from tumor microenvironment (TME) and by diverse autocrine loops. The interplay between tumor cells and their TME during tumorigenesis is likely to induce the phenotypic plasticity that we observe in mesenchymal TNBC tumors.
The Receptor for Advanced Glycation End-product (RAGE) is a multi-ligand member of the immunoglobulin receptor family over-expressed in several pathological conditions involving chronic inflammation. Although RAGE is physiologically involved in the resolution of acute inflammation, clinical studies have linked the expression of RAGE to different types of cancer, diabetes, and neurodegeneration.
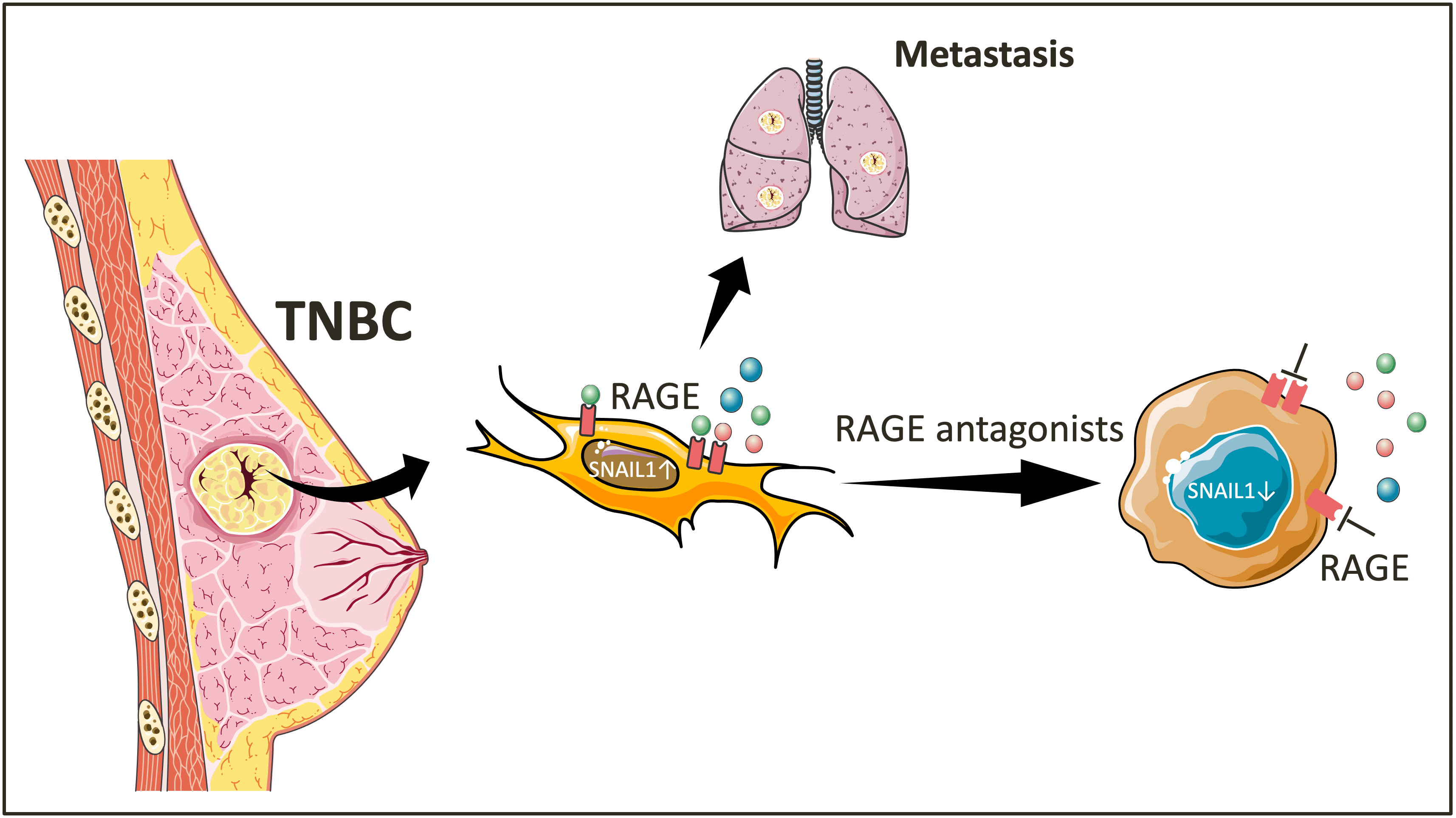 Figure 1. The role of RAGE in TNBC tumor cell plasticity
Figure 1. The role of RAGE in TNBC tumor cell plasticity
A few years ago, careful analysis of TNBC secretomes generated in our lab revealed the existence of several theoretically intracellular proteins. During these studies, we showed that an alternative extracellular function of the nuclear protein HMGA1 mediates tumor invasion and metastasis in triple-negative breast cancer (TNBC) by becoming a ligand for the Receptor for Advanced Glycation End-product (RAGE). Upon these results, we decided to characterize the role of RAGE in TNBC. The main highlights of our work are:
(a) RAGE-SNAIL1 signaling is required to maintain the mesenchymal phenotype of aggressive TNBC cells.
(b) Tumor cell plasticity induced by RAGE is mediated by the EMT transcription factor SNAIL1. Both RAGE antagonists and the ablation of RAGE expression decreases the phosphorylation of ERK1/2 followed by SNAIL1 down-regulation in different TNBC cells.
(c) A cross-talk between RAGE and TGF-β pathways mediates EMT through Smad2/ERK1/2 and SNAIL1. Noteworthy, RAGE antagonists are able to interfere with the induction of EMT by TGF-β1.
(d) Acute acidosis induces an over-secretion of RAGE ligands, increases SNAIL1 levels and tumor cell invasion in a RAGE-dependent manner.
(e) The administration of a RAGE antagonist to TNBC xenograft models reduces metastasis incidence increasing animal survival while downregulating the levels of RAGE and HMGA1 in the invasive front of primary tumors. These results open a new avenue for TNBC therapeutics.
(f) The membrane localization of RAGE is enriched in primary tumors from TNBC patients who later on developed distant metastasis. Therefore, the levels and subcellular localization of HMGA1 and RAGE could predict the incidence of metastasis.
Collectively, all these results reinforce our hypothesis that RAGE plays a key role in the cellular plasticity of mesenchymal TNBC, and suggest that targeting RAGE signaling mediated by extracellular ligands could be an effective approach to fight TNBC.
Follow the Topic
-
Oncogene
This journal aims to make substantial advances in our knowledge of processes that contribute to cancer by publishing outstanding research.





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in