Real-time structure search and structure classification for AlphaFold protein models
Published in Protocols & Methods

Proteins are the fundamental building blocks required for various cellular processes. A protein is comprised of a sequence of amino acids that folds into a 3D structure to perform a specific function, including interaction with other proteins in the cell. Computational prediction of the 3D structure of proteins has been a long-standing challenge that remained unsolved over the past 50 years, where the breakthrough by AlphaFold2 was observed in 2021. AlphaFold2 is a computational protein structure prediction method that showed a substantial improvement in the modeling accuracy, often achieving accuracy expected from experimental methods such as X-ray crystallography.
The impact of AlphaFold2 modeling accuracy is significant as many proteins that do not have experimentally determined structures now have computational models with an expected high accuracy. The database of such computational models was first released with proteomes of 21 species through the AlphaFold Database in July 2021. The release included over 300 thousand structures, which presented a new challenge: Can we facilitate structural analysis of this model by performing a structure-based search? Can we do this structural search in real-time? Answering these questions will provide an invaluable resource for the biology community, which is what we addressed in our Nature Communications Biology paper.
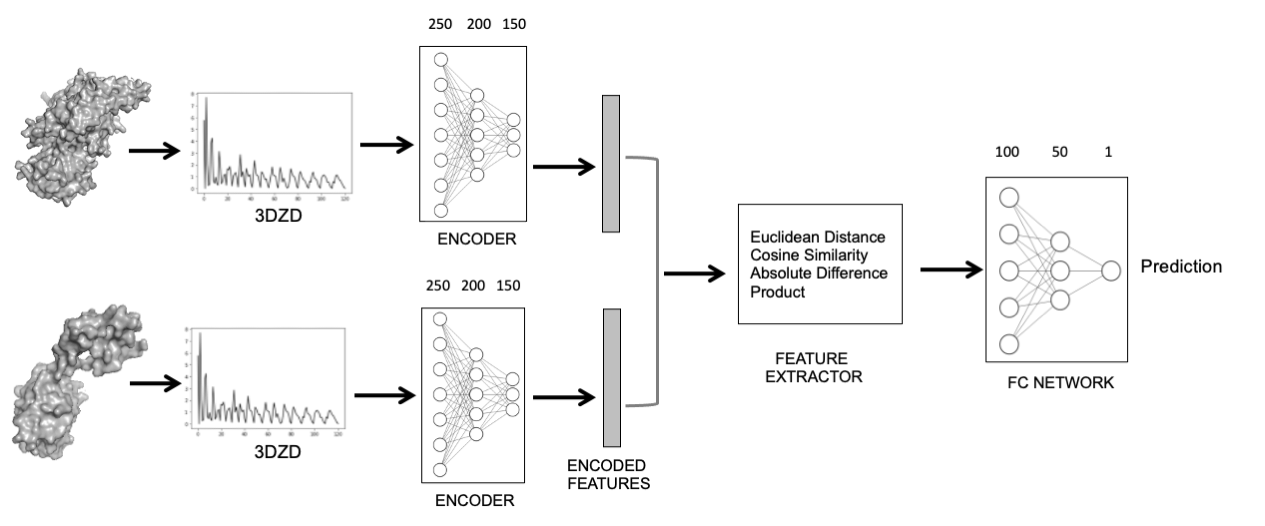 Figure 1: The structure of the neural network borrowed from our nature communications biology paper. The 3DZD representation of the protein structures is fed into the encoder, the embedding output is combined and input into the FC network to predict the similarity of the pair of protein structures. (The figure taken from Figure 3 of our publication)
Figure 1: The structure of the neural network borrowed from our nature communications biology paper. The 3DZD representation of the protein structures is fed into the encoder, the embedding output is combined and input into the FC network to predict the similarity of the pair of protein structures. (The figure taken from Figure 3 of our publication)
Our method, 3D-AF-Surfer, allows real-time structure-based search for AlphaFold2 models and across experimentally determined protein structures in a public database, the Protein Data Bank (PDB). 3D-AF-Surfer represents structures with 3D Zernike descriptors (3DZD), which is a rotationally invariant mathematical representation of 3D shapes. Our lab pioneered this approach of 3D shape representation of protein structure for fast protein similarity comparison. Originally, we compared 3DZD with the Euclidean distance. 3D-AF-Surfer further extends this approach using neural network that produces embeddings of the 3DZD representation of the protein structures.
The performance analysis of 3D-AF-Surfer showed over 40% accuracy improvement over the direct comparison of 3DZD, and it is the only tool that can search between AlphaFold2 models and PDB entries in real-time, within seconds to a couple of minutes. The webserver for performing the real-time structure search is available freely to the public at https://kiharalab.org/3d-surfer/submitalphafold.php
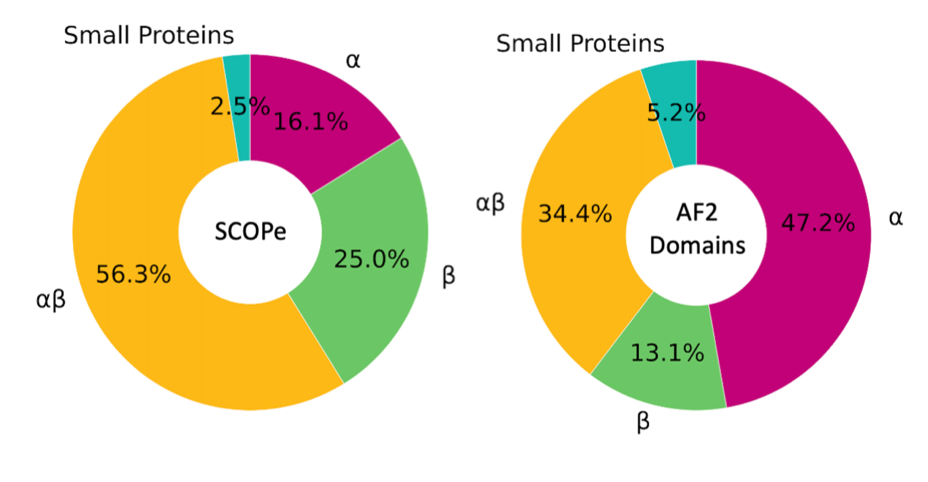 Figure 2: The distribution of the secondary structure classification of AlphaFold2 models compared to experimentally solved structure. Figure taken from Fig. 2A of our publication
Figure 2: The distribution of the secondary structure classification of AlphaFold2 models compared to experimentally solved structure. Figure taken from Fig. 2A of our publication
We used 3D-AF-Surfer to analyze the distribution of the protein folds of Alphafold2 models. Figure 2 shows the distribution of the fold (secondary structure) classes of the Alphafold2 models (the right panel) in comparison with the SCOPe protein structure classification database, which includes protein structures determined by experiments (the left panel). Interestingly, our results showed that the alpha helix class dominates the distribution with over 47% of the structural domains classified as the alpha class. This result contrasts with the distribution observed in experimentally solved structured wherein the alpha/beta class is expected to dominate. We also analyzed unconfident regions of Alphafold2 models in comparison with disordered region prediction from protein sequence. The result showed that at most only 30 to 50%, of unconfident regions correspond to disordered regions, which implies that the rest of the regions would be folded in the native structures. The analysis highlights potential difference of the Alphafold2 models with the native structures, which are yet to be solved.
Finally, we have incorporated newly released predicted models by Alphafold2 into our database, bringing the total number of available domains to 1,315,752 taken from 990,841 proteins. This is 39% increase from the time we wrote the paper. The search speed was not much compromised by this addition. Searching against all the available databases takes about 2.5 minutes.
Follow the Topic
-
Communications Biology

An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.
Related Collections
With Collections, you can get published faster and increase your visibility.
Signalling Pathways of Innate Immunity
Publishing Model: Hybrid
Deadline: Feb 28, 2026
Forces in Cell Biology
Publishing Model: Open Access
Deadline: Apr 30, 2026
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in