
C‒O bonds are an integral part of organic matter and their transformation either via hydrogenolysis or oxidation are fundamental tools to process organic compounds. The predominant paradigm for performing such reactions is based on the use of stoichiometric amounts of sacrificial redox agents which typically include highly reactive and hazardous reagents resulting in several practical challenges ranging from safety hazards over waste management to undesired side reactivity.
Driven by my interest to explore electrophiles as a platform for innovation in catalysis1,2, I asked myself whether the utilization of strategically functionalized C‒O bonds would allow those reactions to be performed in the absence of external redox agents thereby alleviating some of the persisting challenges. Going through the literature, I discovered that the foundation had once again been laid in the allylic realm.3,4 Seminal work by Tsuji, Mandai and co-workers indeed showcased that the use of external reagents could be circumvented by instead harnessing innate C–H bonds of the molecular frame as redox equivalents for the selective transformation of a C–O bond via a hydride transposition akin to traditional transfer (de)hydrogenations. In these pioneering studies the authors demonstrated that, under Pd-catalysis, allyl formates could be used to selectively reduce allylic C–O bonds to the corresponding C–H bonds while allyl carbonates could be used to oxidize aliphatic C–O bonds to the corresponding carbonyls.
Intrigued by this purely catalytic reactivity profile, I set out to build upon these fundamental studies and wondered whether the strategic utilization of a single functional group, which would facilitate reductive as well as oxidative hydride transpositions in a unified manner, could give rise to a general principle for performing mild and selective redox transformations of C–O bonds in the absence of external redox agents, especially targeting the challenging hydrogenolysis of C(sp2)–O bonds. As a result of this design principle, the preinstalled functional group would serve as a redox tag – a synthetic handle dictating the desired redox reactivity of the C–O bond while also allowing for the differentiation between C–O bonds.
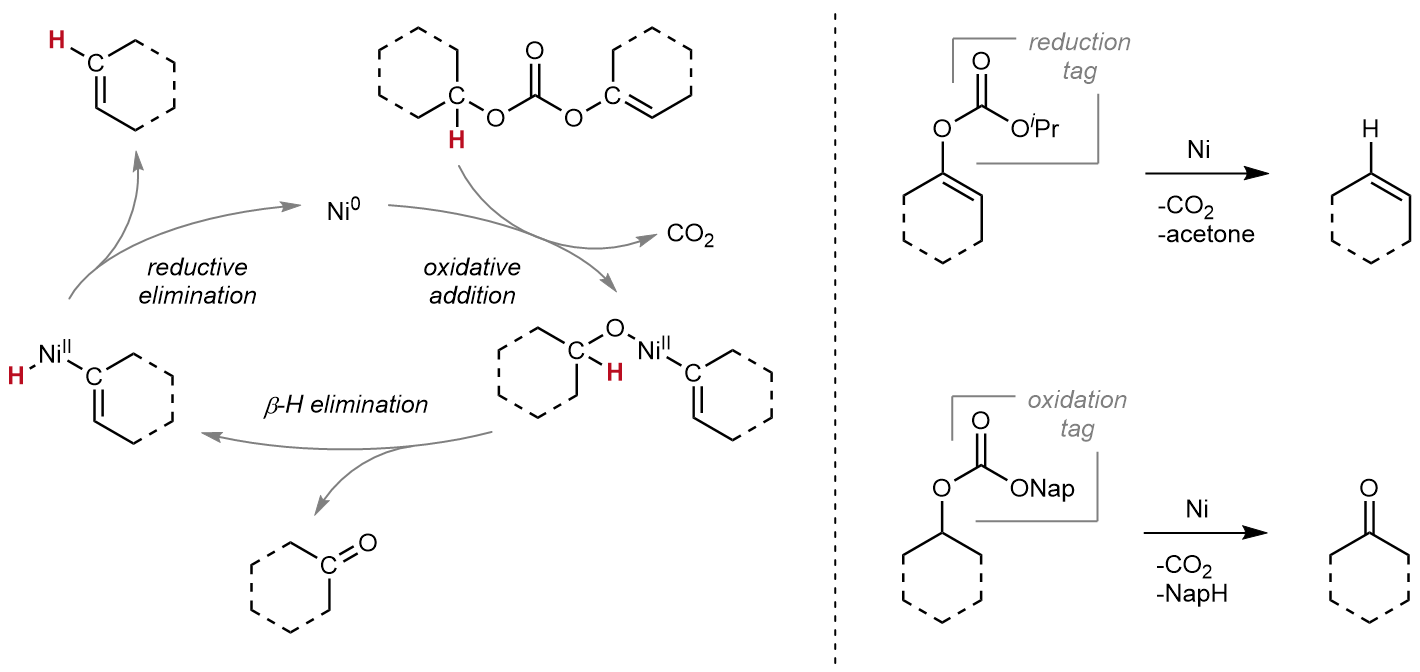
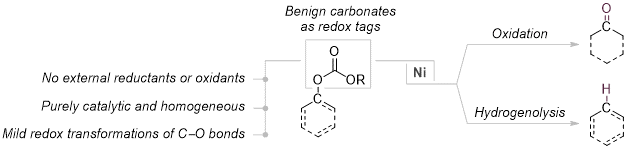
Figure 1 Carbonates as redox tags for the selective, catalytic hydrogenolysis or oxidation of C–O bonds in the absence of external redox agents.
Here, we demonstrate the use of carbonates as versatile redox tags which, in combination with a Ni-catalyst, enable mild hydrogenolysis as well as oxidations of various oxygenated molecules (Figure 1). For example, this purely catalytic strategy allowed for the hydrogenolysis of strong C(sp2)–O bonds down to room temperature in the absence of an external reducing agent or any other additive while traditional protocols, although employing highly reactive and hazardous reductants such as hydrogen or silanes, routinely require temperatures above 100 °C. Besides the mild reaction conditions, the developed redox tag strategy could be readily miniaturized without any adjustments to the reaction conditions and was fully compatible with liquid handling techniques as well as applicable to fluorescence-based reactivity sensing – all representing advantageous parameters for the potential adaptation to emerging technologies such as high-throughput experimentation or flow chemistry. In addition to the synthetic investigation, the work was complemented by a mechanistic study providing detailed insights to the underlying processes. Ultimately, the work aims to highlight the versatile opportunities redox tag strategies offer for organic synthesis and strives to pave the way for future advancements in this field of research.
For more details especially on important prior work please have a look at our article:
https://www.nature.com/articles/s41467-023-38305-y
References
- Toupalas, G. & Morandi, B. Non-innocent electrophiles unlock exogenous base-free coupling reactions. Nat. Catal. 5, 324‒331 (2022).
- Toupalas, G. et al. Pd-Catalyzed Direct Deoxygenative Arylation of Non-π-Extended Benzyl Alcohols with Boronic Acids via Transient Formation of Non-Innocent Isoureas. ACS Catal. 12, 8147‒8154 (2022).
- Mandai, T., Matsumoto, T. & Tsuji, J. A Palladium way to exocyclic methylene by formal anti-thermodynamic isomerization of an endocyclic double bond to an exo position. Synlett 113–114 (1993).
- Minami, I. & Tsuji, J. Dehydrogenation of alcohols with allyl carbonates catalyzed by palladium or ruthenium complexes. Tetrahedron 43, 3903–3915 (1987).
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in