
Drug development is an extraordinarily boundless yet necessary process that takes about a decade from early identification to final FDA approval. The preclinical and clinical phases are crucial in bringing out possible toxicity throwback on the organism [1].
In the last years, the application of artificial intelligence (AI) in drug design and drug discovery has been widely adopted to exploit the big amount of data available to create affordable and reliable models to be effectively used in decision-making [2]. Accordingly, the effort in terms of cost and time in designing a clinical trial could potentially be reduced owing to the employment of AI techniques.
The process that led us to a multi-classifier started from the question of whether or not molecular descriptors could be used to train performant machine learning (ML) models. In our opinion, such an approach could be valuable in supporting results from classical computational methods, e.g., docking and pharmacophore approaches.
In this work, we focused our attention on a specific protein family, the kinase family, that consists of 518 enzymes that regulate almost all aspects of cell life and whose alterations are the cause of cancer and other diseases. The "polypharmacology paradigm" has shown that the treatment of rare cancers is beneficial through the inhibition of multiple protein kinases, especially for cancers where liquid biopsy, which improves the speed and efficiency of diagnosis, allows treatment to begin at an early stage of disease progression.
In addition to cancer, the regulation of kinase function has been shown to play an important role in immunological, inflammatory, degenerative, metabolic, cardiovascular, and infectious diseases [3].
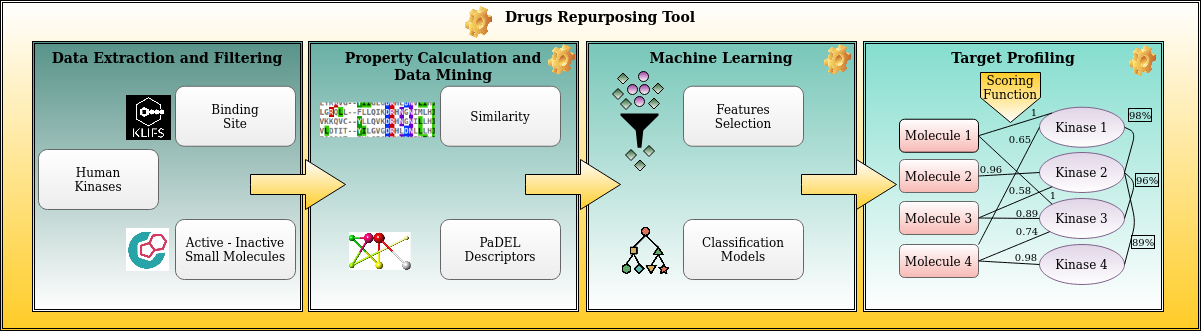
In this context, a novel ML-driven approach, the Kinase drUgs mAchine Learning frAmework (KUALA), has been proposed to reposition known molecules against putative alternative targets belonging to the kinase family. KUALA executes data pre-processing and generates feature selection combinations to find optimal molecular descriptors related to kinase-specific inhibitors (Fig. 1). Finally, the training process leads to the identification of ML models with the best performances to predict the ligand’s activity.

Figure 1. The most frequent descriptors emerged from feature selection used to train the models.
However, it is well-known that many kinase inhibitors can act on different targets simultaneously.
Hence, the identification of inhibitors aims to find the right trade-off between selectivity and modulating multiple targets. For this reason, a multi-target priority score was created and computed by exploiting the similarity between kinase binding sites to prioritize ligands over targets. Here the strategy is the higher the number of molecules predicted as actives for a given kinase, the less selective the model. In Table 1, as an example, we provide a comparison between some structures of predicted actives and known active ligands and/or drugs for some of the kinases.

Table 1. Predicted and known actives for kinases. A comparison of the structures of predicted active ligands for kinases Q13535, Q02750, P31749, P25098, and Q06418 and the structures of some of their known actives.
Moreover, a repurposing threshold was calculated to suggest the molecules most likely to enter drug repurposing.
In order to further investigate the reliability of the KUALA method, validation was also performed by exploiting known drugs. Furthermore, the results of the KUALA approach were also investigated by performing molecular docking, a structure-based approach that predicts binding pose and interactions with a target. The 3D structures with the best resolution of the most representative proteins were downloaded from the Protein Data Bank and used to compare the binding poses of co-crystallized and docked ligands. This analysis showed that most of the crucial interactions and the binding mode were maintained (Fig. 2).
We provided a web portal to browse and download results available at shinyapps.io. Trained models can be downloaded from Zenodo and details about scripts and models are contained in a GitHub repository.

Figure 2. Binding mode of co-crystallized (green) and predicted (orange) ligands.
In conclusion, our work provided a meaningful computational framework able to suggest relevant molecular descriptors associated with kinase activity. The retrieved structural insights such as significant substructures information can help guide drug design campaigns. Moreover, this work allowed us to collect thousands of putative inhibitors that can be used for drug repositioning available in the online repositories.
References
[1] Harrer, S., Shah, P., Antony, B. & Hu, J. Artificial Intelligence for Clinical Trial Design. Trends Pharmacol Sci 40, 577–591 (2019)
[2] Paul, D. et al. Artificial intelligence in drug discovery and development. Drug Discov Today 26, 80–93 (2021).
[3] Ferguson, F. M., & Gray, N. S. (2018). Kinase inhibitors: the road ahead. Nature reviews Drug discovery, 17(5), 353-377.
Follow the Topic
-
Scientific Reports
An open access journal publishing original research from across all areas of the natural sciences, psychology, medicine and engineering.

Related Collections
With Collections, you can get published faster and increase your visibility.
Infectious disease diagnostics
Publishing Model: Open Access
Deadline: Sep 23, 2026
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in