
Why have not more TCRs reactive to shared neoantigens been used in patient treatment?
CD8+ T cells genetically equipped with a therapeutic T cell receptor (TCR) that specifically recognizes a mutated peptide – a neoantigen - expressed by multiple patients, seems like the ideal cancer immunotherapy. Adoptive TCR T cell therapy targeting and eliminating cells expressing hotspot mutations driving cancer could potentially be curative. So why are there only few success stories?
In practice, development of such therapies is challenging for several reasons (Figure 1): First, most mutations in cancer are not shared between patients, but rather unique to the individual cancer patient. Second, many mutations are not presented in the form of mutated peptides on major histocompatibility molecules (MHC) on the surface of cancer cells. Third, among the mutated peptides that are presented on MHC, only a minority is immunogenic, i.e. has the capacity to stimulate T cells. Consequently, finding a TCR that is highly reactive to a shared neoantigen is like finding a needle in a haystack. In general, it has proven difficult to identify patient-derived TCRs that recognize neoantigens with high sensitivity, possibly due to mechanisms of tolerance?

Identification of a FLT3-D835Y-reactive TCR – the needle in the haystack
FLT3 is a gene that is important for the survival and differentiation of normal hematopoietic cells. It is commonly mutated in acute myeloid leukemia (AML) and the FLT3-D835Y mutation is the most frequent point mutation in the intracellular tyrosine kinase domain, making it a potentially attractive target. Using mass spectrometry, we were able to demonstrate that a peptide encoded by the mutation was presented on the surface of patient AML cells in context of the frequent HLA-A*02:01 (HLA-A2) molecule. Having overcome these challenges, the next hurdle was identification of a specific TCR. We could not identify a FLT3-D385Y-reactive TCR among AML patient cells. As the T cells of healthy donors have not undergone thymic negative selection to mutated non-self-peptides, or mechanisms of peripheral tolerance that might exist in AML patients, we hypothesised that they would contain TCRs specific to FLT3-D835Y peptides presented on the frequently expressed HLA-A2 allele. In our study, we used our established technology1,2 to identify neoantigen-specific TCRs from healthy donors. This turned out to be more challenging than we expected. Only after stimulating naïve T cell repertoires from altogether 16 donors, did we identify a TCR with the desired reactivity, demonstrating a very low precursor frequency. Next, we exposed T cells transduced to express the TCR (TCR T cells) to the systematic in vitro safety pipeline that we recently developed3, to accurately map the specificity of the TCR and reveal potential off target reactivities. This screening did not identify cross-reactivities to naturally processed and presented peptides, suggesting that the TCR would not show unintended reactivities to normal cells.
Efficacy of TCR T cells in vitro and in disease-relevant in vivo models
The FLT3-D835Y TCR T cells efficiently killed AML cells isolated from patients carrying both the FLT3-D835Y mutation and the HLA-A2 allele. In contrast, normal leukocytes lacking the FLT3-D835Y mutation were spared, as expected. We wanted, however, extend this finding and explore the efficacy of neoantigen-specific TCR T cells in disease-relevant models, rarely included in such studies. We set up patient-derived xenograft (PDX) models generated from two different patients, each recapitulating a different leukemic phenotype; a very aggressive leukemia, a CD34+ driven leukemogenesis and a minimal residual disease (MRD) setting respectively. After leukemia was established in the PDX mice, we treated them with the FLT3-D835Y TCR T cells or control TCR T cells expressing the clinically applied TCR 1G4, targeting the cancer/testis antigen NY-ESO1. In all our PDX models, we observed an almost complete elimination of the AML cells in the mice treated with FLT3-D835Y TCR T cells, while the leukemic burden remained high or unaffected in the control treated mice. The non-mutated B cells were spared, as expected (Figure 2). As a final model, we investigated if the FLT3-D835Y TCR T cells could eliminate the leukemic cells possessing leukemic stem cell characteristics: i.e. the ability to generate leukemia following transplantation into mice. We co-cultured primary AML cells with FLT3-D835Y TCR T cells or control T cells in vitro for 48h and then transplanted the product into mice, and indeed, only the mice receiving AML cells co-cultured with control, or no T cells, developed leukemia during the 7-month follow-up time.
Implications and future aspects
Chimeric antigen receptor (CAR) T cell therapies have not been approved for treatment of cancer outside of B-cell malignancies. A major reason is lack of cell surface proteins that can be safely targeted. TCRs can, in contrast to CARs, recognize targets independently of subcellular location, dramatically increasing the number of potential therapeutic targets. Despite recent advances in identification and characterisation of cancer-specific TCR T cells, few have, however, made it into the clinic to benefit patients4,5,6. This could potentially be improved by standardizing the preclinical workflow of TCR identification and testing for safety and efficacy. Our study provides proof-of-concept that T cells equipped with TCRs targeting neoantigens encoded by recurrent driver mutations can eliminate AML, including AML cells with characteristics of cancer stem cells. The results are likely relevant for other cancers characterized by recurrent mutations. Use of healthy donor T cells as a rich source of neoantigen-reactive TCRs1,2 allowed us to screen many TCR repertoires to identify a neoantigen-reactive TCR. Application of our recently published systematic safety pipeline3 gave us a map of the TCR fine-specificity and suggested lack of clinically relevant off-target reactivity. Efficacy was tested in multiple PDX models generated from primary patients, which are far more relevant clinically, as compared to xenograft models generated from leukemic cell lines. For instance, the generation of mutated leukemia cells as well as non-mutated B cells from the same patient in one of our PDX models allowed us to assess on and off-target effects of the TCR in one system (Figure 2). Taken together, our proof-of-concept study paves the way for identification of other TCRs targeting recurrent mutations and provides a roadmap for preclinical validation of TCR safety and efficacy, which might ultimately lead to novel and efficient TCR T cell therapies.

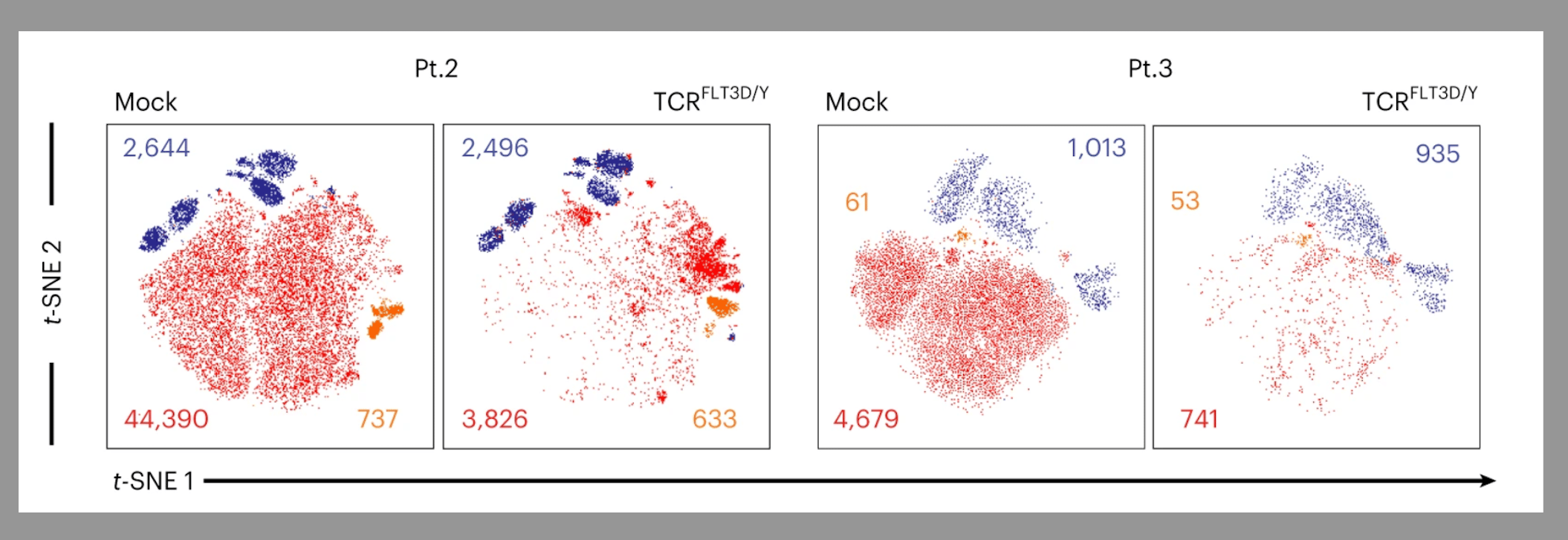
Figure 2. FLT3-D835Y TCR T cells eliminate primary AML cells in vivo. (a) Experimental set up. (b) Flow cytometric profiles of bone marrow (BM) from mice treated with control TCR T cells (top) and FLT3-D835Y TCR T cells (bottom) 34 days after treatment showed complete elimination of the CD34+ AML compartment by the FLT3-D835Y TCR T cells while non-mutated CD19+ B cells were spared.
References:
- Strønen, Toebes, Sander et al., 2016 Jun 10;352(6291):1337-41. doi: 10.1126/science.aaf2288. Epub 2016 May 19.
- Ali, Foldvári, Giannakopoulou et al., Nat Protoc. 2019 Jun;14(6):1926-1943.doi: 10.1038/s41596-019-0170-6. Epub 2019 May 17.
- Foldvári, Knetter, Yang et al., NPJ Vaccines. 2023 Aug 22;8(1):126.doi: 10.1038/s41541-023-00713-y.
- Van der Lee, Reijmers, Honders et al., J Clin Invest. 2019 Feb 1;129(2):774-785. doi: 10.1172/JCI97482.Epub 2019 Jan 14.
- Leidner, Silva, Huang et al., N Engl J Med. 2022 Jun 2;386(22):2112-2119. doi: 10.1056/NEJMoa2119662.
- Kim, Vale, Zacharakis et al., Cancer Immunol Res. 2022 Aug 3;10(8):932-946. doi: 10.1158/2326-6066.CIR-22-0040.
Authored by
Madeleine Lehander, Petter Woll, Sten Eirik Jacobsen and Johanna Olweus
Follow the Topic
-
Nature Cancer
This journal aims to provide a unique forum through which the cancer community will learn about the latest, most significant cancer-related advances across the life, physical, applied and social sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Cancer Neuroscience: from mechanisms to therapy
Publishing Model: Hybrid
Deadline: Jan 30, 2027



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in